Science That Changes Lives
From breakthroughs in the lab to real-world progress—accelerating research that delivers results for families today.
Grant - Winter 2012 - DMD/BMD - Pura Muñoz-Canoves, Ph.D.

MDA awarded a research grant totaling $390,000 over a period of three years to Pura Muñoz-Canoves, ICREA research professor and cell biology coordinator at Pompeu Fabra University in Barcelona. The funds will help support Muñoz-Cánoves' research into strategies aimed at reducing muscle scarring (fibrosis) in people with Duchenne (DMD) and Becker (BMD)muscular dystrophies.
Fibrosis is a hallmark of DMD and a major factor in gauging disease severity. Fibrosis also compromises the efficacy of ongoing preclinical gene- and cell-delivery therapies. No treatment exists for reversing fibrosis in DMD; nor is there a clear understanding of the mechanisms underlying fibrosis development in dystrophic muscle (muscle that is deficient in dystrophin protein).
Muñoz-Canoves and colleagues plan to test whether and how a protein called PAI-1 (plasminogen activator inhibitor-1) may regulate inflammation-driven muscle degeneration and fibrosis development in muscular dystrophy.
"The discovery of new causes of fibrosis development in dystrophic muscle will provide an opportunity for new strategies to halt disease progression," Muñoz-Canoves said. "Since muscle fibrosis also represents a major obstacle for successful engraftment of stem cells in dystrophic muscle, targeting molecules promoting fibrosis appears to be an easy-to-test alternative to improve future DMD stem cell therapies.
Funding for this MDA grant began February 1, 2012.
Grantee: DMD/BMD - Pura Muñoz-Canoves, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2012 - EDMD/LGMD/CMT — Yosef Gruenbaum, Ph.D.

MDA awarded a research grant totaling $300,009 over three years to Yosef Gruenbaum, professor and elected chairman at the Alexander Silberman Institute of Life Sciences, Hebrew University of Jerusalem, in Israel. The funds will help support Gruenbaum’s study of proteins called lamins and their role in muscle diseases such as Emery-Dreifuss (EDMD) and limb-girdle (LGMD) muscular dystrophies, and Charcot-Marie-Tooth disease (CMT).
Relatively little is known about the biological pathways that are affected in muscle cells that have lamin defects, and why these changes lead to muscular dystrophy, Gruenbaum says. Now, using a nematode (worm) model of EDMD, Gruenbaum and colleagues are studying those pathways.
The investigators will use a variety of techniques in structural biology, genetics, cell biology and live imaging to investigate the effects of EDMD-linked lamin mutations and determine how those mutations affect the normal health and development of muscles.
Gruenbaum’s work is expected to uncover the underlying mechanisms at work in EDMD and potentially could lead to the identification of new drug targets and the development of new therapies to treat EDMD.
Funding for this MDA grant began Aug. 1, 2012.
Grantee: EDMD/LGMD/CMT — Yosef Gruenbaum, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD/BMD - Nick Menhart, Ph.D.

Nick Menhart, associate professor of biology at the Illinois Institute of Technology, was awarded an MDA research grant totaling $265,251 over a period of three years to study the properties of modified dystrophin proteins in Duchenne (DMD) and Becker (BMD) muscular dystrophies.
"One of the leading near-term prospects for meaningful treatment of DMD is exon skipping," Menhart said, in which cellular machinery cuts out flawed genetic instructions during protein synthesis, allowing for the production of shortened but at least partially functional dystrophin protein. (Deficient levels of dystrophin protein are the underlying cause of DMD and BMD.)
There are alternative ways to skip portions of the dystrophin gene to effect the repair achieved with exon skipping. Each produces differently modified proteins, having different properties that, in turn, make them better or worse repairs.
Menhart and colleagues plan to characterize different "repaired" versions of dystrophin protein in an attempt to determine which are superior and which will be "maximally effective," in any given person, based on their specific genetic defect.
"This is clearly an exciting and even inspirational time in muscular dystrophy research, as exon skipping holds promise as perhaps a truly effective treatment for DMD," Menhart said. "A number of issues remain in translating this to a clinical setting, but so far … the outlook is good."
Funding for this MDA grant began February 1, 2012.
Grantee: DMD/BMD - Nick Menhart, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – SMA - John Manfredi, Ph.D.

John Manfredi, chief scientific officer at Sfida BioLogic in Salt Lake City, Utah, was awarded an MDA research grant totaling $161,995 over a period of two years to study the potential of new compounds for treatment of spinal muscular atrophy (SMA). The new grant complements previous MDA-funded research by Manfredi into potential therapeutics for SMA.
SMA is caused by the loss of muscle-controlling nerve cells called motor neurons. These neurons have long extensions, called axons, which must remain in communication with muscle in order to promote normal movement. Manfredi and colleagues have identified several small molecules that promote the growth of axons of motor neurons. They have shown that these compounds can improve movement in flies with movement defects, and also have shown their potential in a fish model of SMA. They will now move on to test them in a mouse model of SMA.
“Importantly,” says Manfredi, “previous tests of the compounds in healthy adult rats and mice showed that the chemicals are nontoxic, metabolically stable, capable of entering the central nervous system” and remain active for a long time. All these are important for a compound to become a useful drug. Testing the ability of these compounds as potential treatments for SMA is the next step.
“Given the drug-like characteristics of the compounds, positive results in the SMA mice will nominate the compounds for clinical development,” Manfredi says. They also may shed light on whether these or similar compounds have potential in other diseases of axon degeneration, including amyotrophic lateral sclerosis (ALS) and Charcot-Marie-Tooth (CMT) disease.
Funding for this MDA grant began Feb. 1, 2013.
Grantee: SMA - John Manfredi, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - DMD — Zejing Wang, M.D., Ph.D.

Zejing Wang, associate in clinical research at the Fred Hutchinson Cancer Research Center in Seattle, was awarded an MDA research grant totaling $300,000 over a period of three years to develop gene therapy forDuchenne muscular dystrophy (DMD) — with a focus on treating the heart — in a canine (dog) model of the disease.
DMD is caused by a mutation in the dystrophin gene. Gene therapy in DMD has focused on delivering the gene via a viral vector, or carrier, derived from adeno-associated virus (AAV).
“Despite encouraging results using AAV-mediated delivery of dystrophin into skeletal muscles,” says Wang, “few attempts have been made to genetically treat cardiomyopathy.” Cardiomyopathy, or disease of the heart muscle, remains a major cause of death in DMD, with few treatments available.
Mutations in dystrophin also occur naturally in some dog breeds, making them an important model for studying treatment of the disease.
“Our project will develop AAV-mediated gene therapy for treating cardiomyopathy in a preclinical DMD dog model that can then be applied to treat human DMD patients suffering from cardiac failure. The preclinical DMD dog model represents the most relevant animal model that faithfully mimics human DMD, reproducing many of its features not seen in the mouse model, including cardiomyopathy.”
Wang’s work will focus on improving the ability of the AAV vector to enter heart muscle cells and carry on long-term dystrophin production there.
“Developing strategies for efficient gene delivery and sustained transgene expression in heart muscle will increase the likelihood of achieving the goal of effective gene therapy and the ultimate reduction of mortality in DMD patients,” he says.
Funding for this MDA grant began August 1, 2013.
Grantee: DMD — Zejing Wang, M.D., Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2012 - EDMD — Mary Baylies, Ph.D.
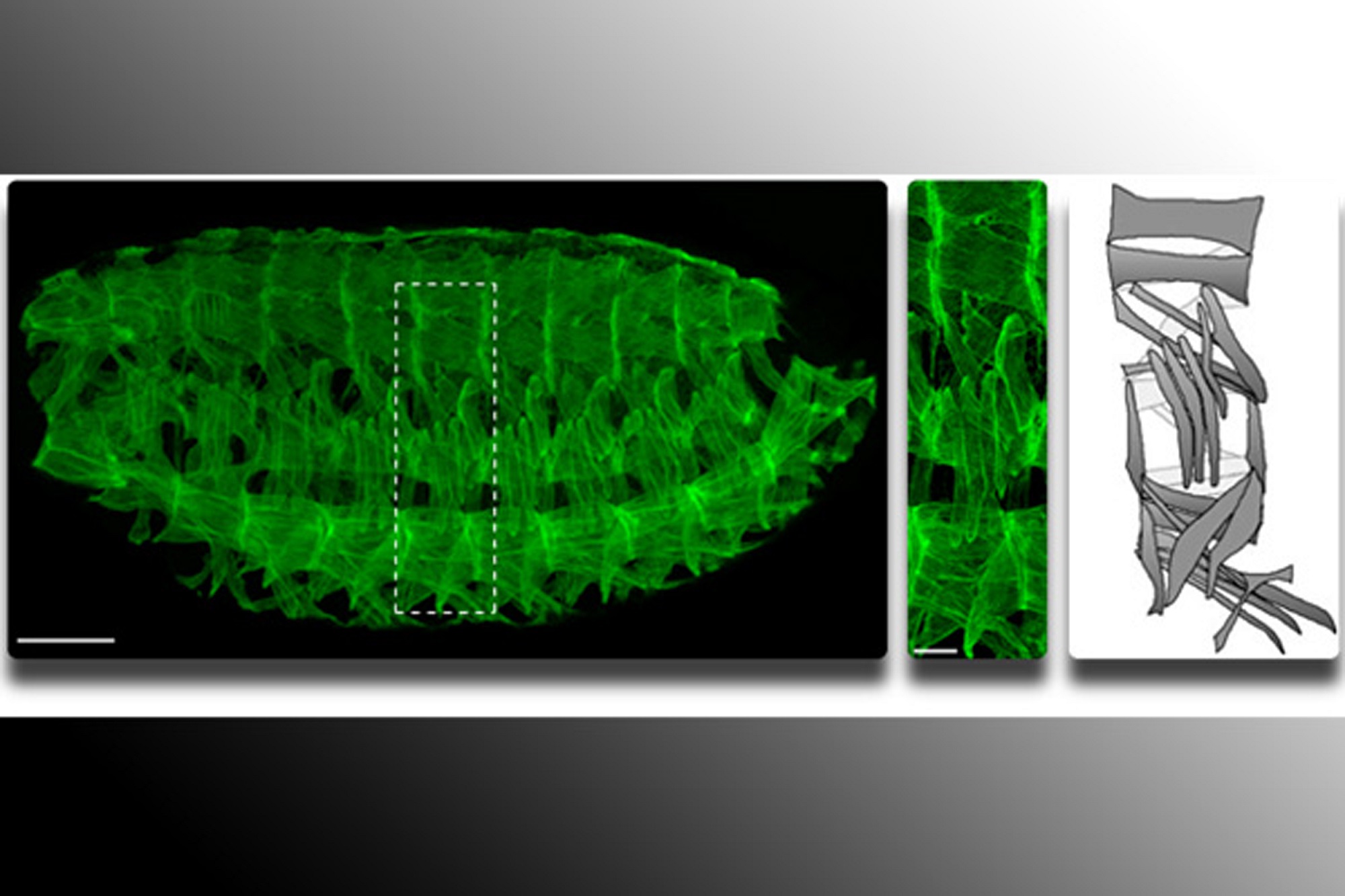
Mary Baylies, professor in the program of developmental biology at Sloan-Kettering Institute, Memorial Sloan-Kettering Cancer Center in New York, was awarded an MDA research grant totaling $399,269 over three years to study LMNA gene mutations and the role of a protein called esconsin in Emery-Dreifuss muscular dystrophy (EDMD).
EDMD can result from mutations in the lamin A/C (LMNA) gene, which carries instructions for the lamin A and lamin C proteins. But how mutations in LMNA cause muscle disease is unclear, Baylies explains.
In previous work, Baylies and colleagues have shown that lamin C interacts with ensconsin, which plays a key role in muscle development.
Now, Baylies is working to determine the nature of the interaction of the two proteins and how they regulate muscle function, with the goal of providing new insight into the cellular processes required for optimal muscle function and for different muscle diseases.
Studies are being conducted in a drosophila (fruit fly) research model, which Baylies says is "particularly well-suited to rapidly finding processes critical for muscle function, due to similarities with mammalian systems at both the genetic and cellular levels." In the model, using time-lapse imaging, Baylies' team gets "an unparalleled view" of cellular processes required for both the development and maintenance of muscle.
“This is an exciting period in muscle biology,” Baylies says. “Imaging approaches continue to evolve and allow us to ‘see’ muscle structure like never before.”
Funding for this MDA grant began Aug. 1, 2012.
Grantee: EDMD — Mary Baylies, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD/BMD - Nadine Wiper-Bergeron, Ph.D.

MDA awarded a research grant totaling $312,422 over a period of three years to Nadine Wiper-Bergeron, assistant professor in the department of cellular and molecular medicine at the University of Ottowa in Ontario, Canada.
The funds will help support Wiper-Bergeron's work on improving muscle stem cell transplantation in Duchenne (DMD) and Becker (BMD) muscular dystrophies.
One potential therapy for DMD and BMD is the use of stem cells to repair disease-damaged muscle. Stem cells can help the damaged muscle become healthy, reversing both muscle weakness and loss of muscle mass.
"So far this approach has not been very successful," Wiper-Bergeron said, "because in addition to repair we need some of the stem cells to live in the muscle all the time, to make more cells that can help with repair and muscle growth."
In previous work, Wiper-Bergeron discovered a new approach to improving muscle stem cell transplantation procedures that involves using a drug to reprogram immature muscle cells called myoblasts back into stem cells before transplantation. Using a research mouse model of DMD, Wiper-Bergeron and colleagues will now test their new approach and compare it to existing strategies.
"It is our aim to make myoblast transplantation more efficient and sustainable long-term by repairing injured muscle and creating a population of healthy stem cells within the muscle," Wiper-Bergeron said. "Successful completion of this project will provide the necessary preclinical data to translate our basic science into the clinic."
Funding for this MDA grant began February 1, 2012.
Grantee: DMD/BMD - Nadine Wiper-Bergeron, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – SMA - Gary Bassell, Ph.D.

Gary Bassell, professor of cell biology and neurology at the Emory University School of Medicine in Atlanta, Ga., was awarded an MDA research grant totaling $405,000 over a period of three years to discover new functions of the SMN protein in spinal muscular atrophy (SMA).
SMA occurs when the gene for SMN (survival motor neuron) is mutated, leaving too little SMN protein to perform its normal functions. While some of those functions are known, Bassell expects there are others that have yet to be described.
SMN binds to a molecule in cells called messenger RNA (mRNA), which carries genetic instructions from the nucleus, where they are stored, to the cytoplasm, where they are used to build proteins. Researchers know that SMN interacts with mRNA in the nucleus, but there is also evidence that the two pair up outside the nucleus, in the cytoplasm, as well. Exactly how SMN functions in this role, and exactly where in the cell the pair are located, is unknown.
“Our central hypothesis is that SMN protein plays a critical role in facilitating regulation of mRNA transport in motor neurons,” Bassell says, particularly the long extensions of these cells called axons, which carry nerve impulses to the muscles.
Bassell will use a mouse model of SMA to study the movements of SMN in cells. Using high-resolution fluorescence microscopy and imaging, he will directly visualize those movements within live nerve axons. His goal is to understand the adverse consequences for mRNA regulation when SMN protein is lost.
“We also will use these state-of-the-art methods to investigate whether specific compounds can correct defects in mRNA regulation in motor neurons from SMA mice,” he says, potentially pointing the way for development of new therapies.
Funding for this MDA grant began Feb. 1, 2013.
Grantee: SMA - Gary Bassell, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD/BMD - Julie Saba, M.D.

Senior scientist Julie Saba, at Children’s Hospital Oakland Research Institute in Oakland, Calif., was awarded an MDA research grant totaling $392,467 over a period of three years. The funds will help support Saba’s research into enhancing muscle regeneration and muscle stem cell functions as a new strategy for treating Duchenne (DMD) and Becker (BMD) muscular dystrophies.
In previous work, Saba and colleagues determined that metabolism of alipid (fat-like substance) called sphingosine-1-phosphate, or S1P, is important in maintaining normal muscle development. S1P stimulates cell signals that promote muscle cell survival and activate muscle stem cells.
The team has found that when S1P levels in mice are decreased using drugs or genetic approaches, a corresponding decrease in muscle stem cell activation and muscle regeneration after injury occurs. The investigators also have determined that S1P signaling and metabolism are activated during muscle injury but may be deficient in muscles affected by MD, thereby contributing to poor muscle regeneration.
“In contrast,” Saba said, “when we use a food-derived small molecule that causes accumulation of S1P, we observe improved muscle regeneration and stem cell functions in a mouse model of MD.”
Such findings suggest that stimulating S1P signaling may improve muscle regeneration and strength in people with MD.
In her new work, Saba will study the effects of modulating S1P as a therapeutic strategy designed to work by activating peoples’ own stem cells to improve muscle regeneration.
“We hope that in the future we may also be able to leverage similar strategies of S1P modulation to improve muscle regeneration in combination with cell therapy approaches that are currently showing limited success,” Saba said.
Funding for this MDA grant began February 1, 2012.
Grantee: DMD/BMD - Julie Saba, M.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – SMA - Christine DiDonato, Ph.D.
Christine DiDonato, assistant professor of pediatrics at Northwestern University in Chicago, Ill., was awarded an MDA research grant totaling $405,000 over a period of three years to test treatment strategies for spinal muscular atrophy (SMA).
SMA is due to the loss of SMN protein, caused by a mutation in the SMN1 gene. Replacing the SMN1 gene, or increasing production from a usually silent "backup" copy of the gene called SMN2, both hold promise for treatment. However, DiDonato says, “it is currently unknown how late in the disease process such therapies can be beneficial, in terms of either improving function or halting disease progression.”
Her research will address that question in a mouse model of the disease by determining the latest time at which SMN protein can be re-introduced and still have effect after disease onset in milder forms of SMA.
In addition, DiDonato says the project “will specifically determine if therapies that only increase SMN within the nervous system can correct all deficits in milder forms of SMA. This research has important implications for SMA therapy development,” since diagnosis of the disease often occurs months or years after birth.
DiDonato will measure strength, endurance and gait in mice that begin SMN protein production at various time points. Using these techniques, she will ask “whether we can halt, slow or reverse disease progression when SMN is returned after symptoms are clearly evident, and at advancing points of disease.”
She notes that “great strides have been made in SMA research and therapeutic development. There are now several drugs that have entered clinical trials for SMA, so it is a very exciting time. Our research will provide important information for SMN-based therapies for milder forms of SMA.”
Funding for this MDA grant began Feb. 1, 2013.
Grantee: SMA - Christine DiDonato, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD/BMD - James Ervasti, Ph.D.

James Ervasti, professor of biochemistry, molecular biology & biophysics at the University of Minnesota in Minneapolis, was awarded an MDA research grant totaling $390,000 over a period of three years to help support his research into improving two therapies currently in development for people with Duchenne (DMD) and Becker (BMD) muscular dystrophies.
Exon skipping is a strategy in which molecules called anstisense oligonucleotides (AONs) target error-containing parts of a gene and coax cells to retain the error-free parts for protein synthesis.
As with exon skipping, viral delivery of miniaturized dystrophin genes is designed to restore sufficient levels of the dystrophin protein, which is deficient in DMD and BMD.
Both strategies create non-natural versions of dystrophin, and Ervasti and colleagues have shown that the miniaturized dystrophin proteins are prone to misfolding, instability and clumping into aggregates — all of which likely limit their effectiveness.
In his new research, Ervasti will study ways to assess and optimize the stability of miniaturized dystrophin proteins in the laboratory before they are tested in animal models with dystrophin deficiency. Ervasti's team also will examine how exon-skipping strategies currently under investigation to treat DMD and gene deletions associated with BMD affect the stability of dystrophin.
A better understanding of how missing stretches of the dystrophin protein affect folding "may further allow us to identify drugs to treat patients with BMD and also enhance the effectiveness of exon skipping approaches," Ervasti said.
Funding for this MDA grant began February 1, 2012.
Grantee: DMD/BMD - James Ervasti, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2012 - DMD/BMD — Veronica Hinton, Ph.D.

Veronica Hinton, associate professor of clinical neuropsychology at Columbia University in New York, was awarded an MDA research grant totaling $397,596 over three years to study cognitive problems in children with dystrophinopathies, which include Duchenne (DMD) and Becker (BMD) muscular dystrophies.
"Children with dystrophinopathies are at risk for having delayed language development, poor academic achievement and limited social skills in addition to muscle weakness,” Hinton notes. “This cognitive and behavioral profile puts them at increased risk for poor quality of life, but the profile can be modified and improved with early detection and proper intervention.”
Hinton is working to better understand the specific nature of the cognitive problems that children with dystrophinopathies may experience — information that is vital to minimizing and ameliorating the learning and behavior challenges that many children and families experience.
With colleagues, Hinton is using neuropsychological tests to examine cognitive and behavioral performance in a diverse group of children. The children participate in tests that involve answering questions, solving picture riddles and doing computer-administered puzzle tasks. Parents and teachers complete rating forms about each child’s daily life behavior.
Hinton’s study is designed to do an in-depth assessment of executive function (the higher thought processes that include making or following complicated plans, solving complex problems, following a series of directions and making sound judgments) and verbal working memory skills, and provide information about how these skills affect “real life” outcomes in school, socialization and quality of life.
"Research in this area has been limited, yet effective in raising awareness and understanding about the cognitive and behavioral risks associated with a diagnosis of dystrophinopathy,” Hinton says. “With that knowledge has come much improved care for the individuals affected.”
Funding for this MDA grant began Aug. 1, 2012.
Grantee: DMD/BMD — Veronica Hinton, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – OPMD - Sarah Youssof, M.D.
Sarah Youssof, at the University of New Mexico Health Sciences Center in Albuquerque, was awarded an MDA clinical research training grant totaling $180,000 over a period of two years to study outcome measures in oculopharyngeal muscular dystrophy (OPMD).
OPMD is a progressive, adult-onset disease that affects the muscles that control the eyelids, the swallowing muscles and other muscles. It typically occurs in the fourth or fifth decade of life. The gene that, when defective, causes OPMD encodes a protein called nuclear poly (A) binding protein 1 (PABPN1).
A critical barrier to the pursuit of clinical trials is the lack of established outcome measures that can capture disease progression and treatment effects.
Youssof’s goal is to explore the performance of a set of outcome measures for measurement of OPMD disease severity and to investigate patients’ perspectives on the disease.
The results of Youssef’s work could lead to clinical trials for OPMD incorporating validated outcome measures that reflect endpoints that are meaningful to people with the disease.
Funding for this MDA grant is effective July 1, 2013.
Grantee: OPMD - Sarah Youssof, M.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD/BMD - David Thomas, Ph.D.

MDA awarded a research grant totaling $390,000 over a period of three years to David Thomas, the William F. Dietrich Professor of Structural Biology and Biophysics at the Minnesota Muscle Laboratory, University of Minnesota in Minneapolis.
The funds will help support Thomas' research into the role of calcium trafficking in Duchenne (DMD) and Becker (BMD) muscular dystrophies.
Defects in the sarcolemma (the membrane that surrounds a muscle fiber) of people with muscular dystrophy lead to elevated concentrations of calcium in the muscle fiber — a condition that can cause cell death.
Recent reports have indicated that increasing activity in skeletal muscle of a protein called SERCA increases calcium transport out of the fiber, lessening disease severity in mouse models of muscular dystrophy. Such an approach relies on gene therapy.
Thomas and colleagues plan to develop a small-molecule approach to achieve the same end. The group intends to identify small-molecule compounds that directly activate SERCA from skeletal muscle and then evaluate each compound's ability to restore health and function to single skeletal muscle cells and intact skeletal muscles. Finally, the investigators plan to begin testing the most promising compounds in mice.
The results of this project are expected to provide proof-of-concept of for a small-molecule strategy to treat muscular dystrophy and identify a set of lead compounds for drug development.
Funding for this MDA grant began February 1, 2012.
Grantee: DMD/BMD - David Thomas, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – OPMD - Ayan Banerjee, Ph.D.

Ayan Banerjee, a postdoctoral researcher in biochemistry at Emory University in Atlanta, Ga., was awarded an MDA development grant totaling $180,000 over a period of three years to study the protein defects that cause oculopharyngeal muscular dystrophy (OPMD).
OPMD affects the muscles that control the eyelids, the swallowing muscles and other muscles. It typically occurs in the fourth or fifth decade of life. The gene that, when defective, causes OPMD encodes a protein called nuclear poly (A) binding protein 1 (PABPN1).
“Although we know that the PABPN1 gene is altered in this disease,” says Banerjee, “we do not understand why this change causes a muscle disease, and we also do not currently have any treatment for this disease.”
Banerjee’s goal is to better understand how the PABPN1 protein is regulated, in order to develop a better understanding of how the gene mutation causes disease. His work will include experiments in cell culture and in a fly model of OPMD. “The fly model is very useful for these studies because we can analyze PABPN1 function in the muscle of a living organism quickly and with limited cost, as compared to genetic mouse models,” he says.
Banerjee will be focusing on structural changes made to the protein after it is created (called post-translational modifications), which are known to help regulate the function of the final protein. One of these modifications, called phosphorylation, may be especially important, as errors in phosphorylation are associated with other forms of muscular dystrophy.
“Drugs correcting these defects result in an improvement in the disease in animal models,” Banerjee points out, suggesting a possible therapeutic target for OPMD as well. “If we can understand how the function of PABPN1 can be modulated, we may be able to develop new therapeutic approaches to treat OPMD.”
Funding for this MDA grant began Feb. 1, 2013.
Grantee: OPMD - Ayan Banerjee, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2012 - DMD/BMD — Tom Thompson, Ph.D.
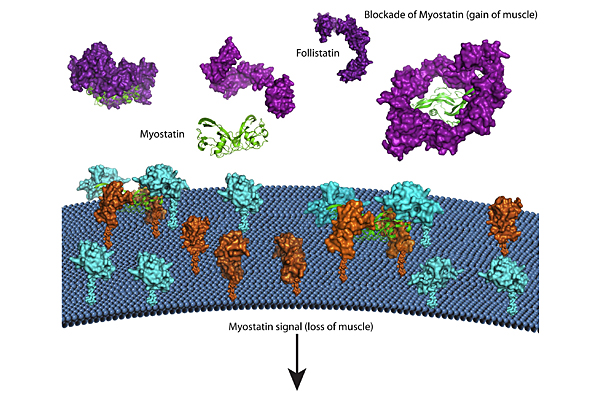
Tom Thompson, associate professor in the department of molecular genetics, biochemistry and microbiology at the University of Cincinnati in Ohio, was awarded an MDA research grant totaling $330,000 over three years. The funds will help support Thompson's study of potential therapies based on blocking a protein called myostatin for Duchenne (DMD) and Becker (BMD) muscular dystrophies.
"In our bodies, a protein called myostatin inhibits the size and mass of muscle, so therapies aimed at inhibition of myostatin are in high demand," Thompson explains. One potential concern, however, is that myostatin inhibitors need to be specific in order to limit potential side effects.
Humans have proteins that naturally inhibit myostatin. Thompson and colleagues are using a technique called X-ray crystallography to determine, at the atomic level, how these naturally occurring inhibitors bind to and neutralize myostatin. Their focus is on a protein called GASP1, which binds myostatin with high specificity.
The investigators plan to identify the mechanism by which GASP1 binds to myostatin.
In addition, the team will study another inhibitor called follistatin, which helps degrade myostatin by binding with cell surface molecules calledheparin.
This knowledge may make it possible to leverage the body's own system to inhibit myostatin, or develop other strategies to block it, Thompson says.
Funding for this MDA grant began Aug. 1, 2012.
Grantee: DMD/BMD — Tom Thompson, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Spring 2014 - SMA - Stephen Kolb, M.D., Ph.D.

Stephen Kolb, an assistant professor in the Departments of Neurology and of Molecular & Cellular Biochemistry at Ohio State University, has been awarded an MDA human clinical trial grant totaling $183,354 over three years as supplemental funding for the SMA NeuroNEXT biomarkers study. This study is being conducted by the U.S. National Institutes of Health (NIH) to compare children with and without spinal muscular atrophy (SMA) during the first two years of life. Its purpose is to determine measurements of SMA progression during this period so that these measurements (“biomarkers”) can be used to assess the effects of experimental treatments in future trials. The MDA funding will allow additional training of evaluators of motor function.
Funding for this MDA human clinical trial grant began June 1, 2014.
Grantee: SMA - Stephen Kolb, M.D., Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD - Vladimir Ljubicic, Ph.D.

Vladimir Ljubicic, a postdoctoral fellow in the department of cellular and molecular medicine at the University of Ottawa in Ontario, Canada, was awarded an MDA development grant (DG) totaling $180,000 over a period of three years for his investigation into potential therapies for Duchenne muscular dystrophy (DMD). (MDA development grants are awarded to exceptional postdoctoral candidates who have the best chance of becoming independent researchers and future leaders of neuromuscular disease research.)
Scientists have shown that increasing levels of a structural protein calledutrophin and properly localizing it in the cell appears to partially compensate for the missing dystrophin protein — the underlying cause of DMD.
Utrophin has been observed at higher levels in "slow-twitch" muscle fibers (which are less affected in DMD), and at lower levels in "fast-twitch" fibers.
"A critical question," Ljubicic said, "is whether the benefits associated with slow-twitch fibers in DMD are strictly dependent on the increased levels of utrophin, or on some other factor associated with the slow-twitch fiber type."
In human skeletal muscle cell cultures, and also in mouse models of DMD, Ljubicic will test several compounds to determine whether they promote development of slow-twitch fibers.
Because the compounds he plans to test already are in clinical trials for other disorders, favorable results from Ljubicic's research could accelerate the development and implementation of new therapies for DMD centered either on increased utrophin activity in the cell and/or promotion of the slow-twitch muscle-fiber type.
Funding for this MDA grant began February 1, 2012.
Grantee: DMD - Vladimir Ljubicic, Ph.D.
Grant type: Development Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – Muscle Physiology - Masahiro Iwamoto, Ph.D., D.D.S.

Masahiro Iwamoto, research scientist at the Children’s Hospital of Philadelphia and associate professor of pediatric orthopedics at the University of Pennsylvania School of Medicine, was awarded an MDA research grant totaling $405,000 over a period of three years to study new ways to reduce muscle degeneration.
“The active form of vitamin A, called retinoic acid, plays an important role in numerous organs, including musculoskeletal systems,” says Iwamoto. His team has recently discovered that a molecule similar to retinoic acid, called an RAR-gamma agonist, blocks two different pathologic processes in muscle: formation of bone within the muscle and muscle degeneration.
Iwamoto will now build on that work to learn more about RAR-gamma signaling and how it contributes to the repair and maintenance of skeletal muscle. He will test whether a specific RAR-gamma agonist, called R667, which has shown some promise in the lab and has been tested in humans for other conditions, has potential for treatment of muscular dystrophy in mice.
“We will determine whether these drugs can in fact block or even reverse muscle degeneration in mouse models of muscular dystrophy,” he says, potentially leading to a novel means of therapeutic intervention.
Funding for this MDA grant began Feb. 1, 2013.
Grantee: Muscle Physiology - Masahiro Iwamoto, Ph.D., D.D.S.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Sping 2014 - SMA - Rashmi Kothary, Ph.D.

Rashmi Kothary, a senior scientist in the Regenerative Medicine Program at Ottawa Hospital Research Institute in Canada, has been awarded an MDA research grant totaling $253,800 over three years to further investigate a potential new treatment approach for spinal muscular atrophy (SMA) . Kothary and colleagues will continue to develop inhibitors of an enzyme called rho kinase to see if they are beneficial in this disease. Early experiments in mice have shown promise.
Funding for this MDA research grant began May 1, 2014.
Grantee: SMA - Rashmi Kothary, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD - Robert Korneluk, Ph.D.

MDA awarded a research grant totaling $425,952 over a period of three years to Robert Korneluk, director of the Apoptosis Research Centre at the Children’s Hospital of Eastern Ontario Research Institute, and distinguished professor at the University of Ottawa in Ontario, Canada.
The funds will help support Korneluk’s study of the Nuclear Factor kappaB(NFkB) signaling pathway in muscle diseases such as Duchenne muscular dystrophy.
The NFkB signaling pathway is critical for normal skeletal muscle function, and is important for promoting recovery in response to muscle injury. There also is strong evidence that NFkB signaling is involved in the disease process of many muscle diseases.
Korneluk and colleagues recently found that two proteins called cIAP1 and cIAP2, initially identified by the group, are required for various aspects of NFkB signaling in skeletal muscle. The investigators also found that the loss of cIAP1 protein activity in muscle, in cultured cells and in mice, leads to changes in the NFkB signaling pathway, with subsequent improvement of muscle function and recovery.
Korneluk’s team plans to define the roles and mechanism of action of cIAP1 and cIAP2 in NFkB signaling in skeletal muscle, and evaluate their contribution to the DMD disease process. Additionally, the investigators will evaluate the therapeutic potential in muscular dystrophy of several drugs that specifically and strongly target cIAP1 and cIAP2 for destruction.
Funding for this MDA grant began February 1, 2012.
Grantee: DMD - Robert Korneluk, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2012 - DMD/BMD — Ryan Wuebbles, Ph.D.

Ryan Wuebbles, a postdoctoral fellow in pharmacology at the University of Nevada School of Medicine in Reno, was awarded an MDA development grant (DG) totaling $180,000 over three years to study the potential use of a protein called laminin-111 as the basis of therapies for Duchenne (DMD) and Becker (BMD) muscular dystrophies.
MDA development grants are awarded to exceptional postdoctoral candidates who have the best chance of becoming independent researchers and future leaders of neuromuscular disease research.
In a research mouse model with a disease resembling DMD, treatment with laminin-111 has been shown to prevent muscle damage and improve resistance to exercise-induced muscle injury. The proteins' large size, however, makes it difficult to manufacture, so Wuebbles and colleagues are working to determine whether a small part of the protein is capable of producing the same effects.
Wuebbles plans to test different parts of laminin-111 in cells cultured from DMD research mice and from people with the disease. The most promising of these will be tested in a DMD research mouse model, where any effects can be compared to those generated by the full-length protein.
"Protein therapeutics are emerging as some of the strongest Duchenne muscular dystrophy therapeutic candidates," Wuebbles says. "Exciting new protein treatments such as laminin-111 offer a directed natural approach compared to traditional small chemical compounds."
Funding for this MDA grant began Aug. 1, 2012.
Grantee: DMD/BMD — Ryan Wuebbles, Ph.D.
Grant type: Development Grant
Award total:
Institution:
Country:
Grant - Winter 2012 - DMD - Lawrence Steinman, M.D.

MDA awarded a research grant totaling $308,061 over a period of three years to Lawrence Steinman, Zimmerman Chair and professor of neurology, neurosciences and pediatrics at Stanford University School of Medicine in Stanford, Calif. The funds will help support Steinman’s work to inhibit immune system response to replacement of the missing dystrophin protein in Duchenne muscular dystrophy.
Dystrophin-specific immunity has been reported in people with DMD both before and after treatment with gene therapy.
Steinman and colleagues plan to “map the exact nature” of the immune response to dystrophin in boys with Duchenne MD. In addition, the investigators have invented a strategy designed to shut down the immune response to dystrophin, and plan “to test this approach in experimental gene therapy in mice with a DMD-like disease.”
Steinman said that most attempts to suppress unwanted immunity in DMD use “big-hammer drugs” with strong immune system suppression properties. His new approach is more selective, he noted, designed to “turn off the immune response specifically when it is unwanted,” leaving the rest of the immune system intact.”
“Ultimately, this could increase the success of gene therapy in DMD,” Steinman said, “by inhibiting an unwanted immune system response when the normal dystrophin molecule is produced.”
Funding for this MDA grant began February 1, 2012.
Grantee: DMD - Lawrence Steinman, M.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2013 – MM – Mitochondrial Myopathies - Eric Schon, Ph.D.

Eric Schon, professor of neurology at the College of Physicians & Surgeons of Columbia University in New York, was awarded an MDA research grant totaling $405,000 over a period of three years to develop strategies for treatment of mitochondrial myopathies.
Mitochondrial myopathies are muscle diseases caused by defects in the mitochondria, subcellular structures that create energy from cellular fuel. Mitochondria contain their own DNA (mtDNA) and reproduce themselves within cells. In people with a mitochondrial myopathy, some mitochondria are mutated and function poorly, while others are healthy.
“There are no general therapeutic strategies to combat diseases involving mitochondrial DNA mutations,” Schon says. His group recently showed that in cells containing only mutated mtDNAs, it was possible to eliminate those mitochondria using rapamycin, a drug that stimulates the cell to degrade defective mitochondria. “We have now found that in cells containing a mixture of normal and mutated mitochondria, rapamycin dramatically increases the proportion of ‘good’ mitochondria and restores cellular bioenergetic function within only a few days.”
The implication of this finding is that inducing cells to selectively degrade their mutant mitochondria, paving the way for more reproduction by the good mitichondria, could be a promising method to treat mitochondrial myopathies.
Schon will explore this potential and study its mechanism, as well as search for other compounds with similar effects. “Using these approaches, we hope to gain insight, both practical and basic, into novel approaches to treat mitochondrial myopathies,” he says.
Funding for this MDA grant began Feb. 1, 2013.
Grantee: MM – Mitochondrial Myopathies - Eric Schon, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Spring 2014 - SMA - Lyndsay Murray, Ph.D.

Lyndsay Murray, a lecturer in anatomy at the University of Edinburgh in Scotland, has been awarded an MDA development grant totaling $152,280 over three years to determine the earliest changes in gene activity that occur in spinal muscular atrophy (SMA). By conducting experiments in mice with and without an SMA-like disorder, Murray and colleagues will study the genetic changes that occur prior to the death of nerve cells in the SMA-like condition. The work is expected to lead to new ideas of how to protect nerve cells that carry SMA-causing abnormalities.
Funding for this MDA development grant began May 1, 2014.
Grantee: SMA - Lyndsay Murray, Ph.D.
Grant type:
Award total:
Institution:
Country:
Find MDA
in your Community
-

Grants at a Glance
Read More -

Search for Clinical Trials
Learn More

