Spinal Muscular Atrophy (SMA)
Spinal Muscular Atrophy (SMA)
Table of Contents
- What is spinal muscular atrophy (SMA)?
- What causes SMA?
- What are the symptoms of SMA?
- What is the progression of SMA?
- What is the status of research on SMA?
- Additional reading
- In the News
What is spinal muscular atrophy?

Spinal muscular atrophy (SMA) is a genetic disease affecting the central nervous system, peripheral nervous system, and voluntary muscle movement (skeletal muscle).
Most of the nerve cells that control muscles are located in the spinal cord, which accounts for the word spinal in the name of the disease. SMA is muscular because its primary effect is on muscles, which don’t receive signals from these nerve cells. Atrophy is the medical term for getting smaller, which is what generally happens to muscles when they’re not stimulated by nerve cells.
SMA involves the loss of nerve cells called motor neurons in the spinal cord and is classified as a motor neuron disease.
In the most common form of SMA (chromosome 5-related SMA, or SMN-related SMA), there is wide variability in age of onset, symptoms, and rate of progression. In order to account for these differences, chromosome 5-related SMA is typically classified into types 1 through 4.
The age at which SMA symptoms begin roughly correlates with the degree to which motor function is affected: The earlier the age of onset, the greater the impact on motor function. Children who display symptoms at birth or in infancy typically have the lowest level of functioning (type 1). Later-onset SMA with a less severe course (types 2 and 3, and in teens or adults, type 4) generally correlates with increasingly higher levels of motor function.
For more, see Types of SMA.
What causes SMA?
Chromosome 5-related SMA is caused by a deficiency of a motor neuron protein called SMN, for “survival of motor neuron.” This protein, as its name implies, seems to be necessary for normal motor neuron function. SMN plays a pivotal role in gene expression in motor neurons. Its deficiency is caused by genetic flaws (mutations) on chromosome 5 in a gene called SMN1. The most common mutation in the SMN1 gene within patients diagnosed with SMA is a deletion of a whole segment, called exon 7. Neighboring SMN2 genes can in part compensate for nonfunctional SMN1 genes as there is 99% identity between these two genes.
Other rare forms of SMA (non-chromosome 5) are caused by mutations in genes other than SMN1.
For more information including detailed reading on rare, non-chromosome 5-related SMA, see Types of SMA and Causes/Inheritance.
What are the symptoms of SMA?
SMA symptoms cover a broad spectrum, ranging from mild to severe.
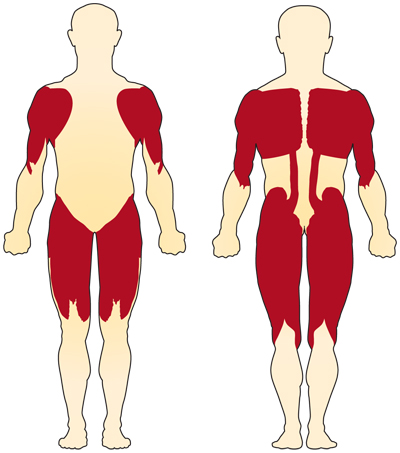
The primary symptom of chromosome 5-related (SMN-related) SMA is weakness of the voluntary muscles. The muscles most affected are those closest to the center of the body, such as those of the shoulders, hips, thighs, and upper back. The lower limbs seem to be affected more than the upper limbs, and deep tendon reflexes are decreased.
Special complications occur if the muscles used for breathing and swallowing are affected, resulting in abnormalities in these functions. If the muscles of the back weaken, spinal curvatures can develop.
There's a great deal of variation in the age of onset and level of motor function achieved in chromosome 5-related SMA. These characteristics are roughly correlated with how much functional SMN protein is present in the motor neurons, which in turn correlates with how many copies of SMN2 genes a person has. Sensory, mental, and emotional functioning are entirely normal in chromosome 5-related SMA.
Some forms of SMA are not linked to chromosome 5 or SMN deficiency. These forms vary greatly in severity and in the muscles most affected. While most forms, like the chromosome 5-related form, affect mostly the proximal muscles, other forms exist that affect mostly the distal muscles (those farther away from the body’s center) — at least in the beginning.
For more, see Signs and Symptoms.
What is the progression of SMA?
In chromosome 5-related SMA, the later the symptoms begin and the more SMN protein there is, the milder the course of the disease is likely to be. While in the past, infants with SMA typically did not survive more than two years, with new therapies and earlier diagnosis, the typical progression of disease is changing. Today, most doctors consider SMN-related SMA to be a continuum and prefer not to make rigid predictions about life expectancy or weakness based strictly on age of onset.
SMA is the most common genetic cause of mortality in infants.
What is the status of research on SMA?
Research has focused on strategies to increase the body's production of the SMN protein lacking in the chromosome 5-related forms of the disease. Approaches include methods to help motor neurons survive in adverse circumstances.
For more, see Research.
To read more about SMA, see:
Additional reading
- Prior TW, Leach ME, Finanger EL. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2024 Sep 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352/
In the News
- Health Day (August 11, 2025) "What You Should Know About Spinal Muscular Atrophy (SMA)"
Last reviewed July 2025 by Aravindhan Veerapandiyan, MD
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Grants at a Glance
Read More -

Search for Clinical Trials
Learn More

