Myotonic Dystrophy (DM)
Adult-Onset DM1 and DM2
The classic form of DM1 becomes symptomatic between the second and fourth decades of life. In these patients, average lifespan is reduced. Patients diagnosed with DM1 have multiple sets of DNA bases repeats in their genome (known as the CTG repeats). The CTG repeat size in adult onset is generally in the range of 50 to 1,000.1
The mild form of DM1 is characterized by mild weakness, myotonia, and cataracts. Age at onset is between 20 and 70 years (typically onset occurs after age 40), and life expectancy is normal. The CTG repeat size is usually in the range of 50 to 150.1
Onset for DM2 ranges from the second to the seventh decade of life, often presenting with myotonia, weakness, or cataracts. In general, DM2 is a less severe disease than classic DM1. In most cases, weakness predominantly involves the proximal muscles, particularly the hip girdle muscles.2
Effects on the brain
Research suggests that, in DM1, there may be abnormalities in the parts of the brain that determine the rhythm of sleeping and waking, making excessive daytime sleepiness a barrier to full participation in work, school, or social life for many adults with the disorder. In some people, there is a kind of overall "apathy" that may be due to changes in the brain related to DM1. Also, in patients with DM1, cognitive skills are diminished, and the IQ has been shown to be lower with younger age of onset. In both classic DM1 and DM2, frontal lobe cognitive impairment (attention deficit) worsens over time but does not extend to other areas of cognition. Thus, cognitive problems do not show the same degree of deterioration over time that is typical of muscle dysfunction in DM1.
Although not as much is known about the effects of DM2 on personality, cognition, and sleepiness as with DM1, it appears that people with DM2 can have some of the same difficulties in these areas but to a lesser degree. Intellectual disability is rare in DM2.
To learn more, read The Brain in DM (cognitive and emotional aspects of DM1) and Excessive Daytime Sleepiness Can Be 'Debilitating' in DM1 and DM2 (complex effects of DM on the brain's sleep-wake cycles and respiratory muscles).
Breathing and swallowing muscle weakness
Respiratory muscle weakness does not appear to be a common feature of DM2. However, in DM1, respiratory muscle weakness can affect lung function and deprive the body of needed oxygen. Weakness of the diaphragm and other breathing muscles can lead to problems getting enough oxygen when a person is asleep, even if they do not have any symptoms of breathing difficulty while awake. Thus, respiratory problems in DM1 can lead to a condition known as sleep apnea, in which people stop breathing for several seconds or longer many times a night while asleep.
Swallowing muscles, if weakened, can lead to choking or “swallowing the wrong way” (called aspiration), with food or liquid going down the trachea (windpipe) to the lungs instead of down the esophagus to the stomach. Swallowing is partly voluntary and partly involuntary, and both voluntary and involuntary muscles can be affected.
Respiratory failure may occur, sometimes precipitated by general anesthesia because of heightened sensitivity to sedatives, anesthetics, and neuromuscular blocking agents.
To learn more, read Excessive Daytime Sleepiness Can Be 'Debilitating' in DM1 and DM2.
Cataracts
Cataracts — cloudy areas of the lens of the eye that eventually can interfere with vision — are extremely common in both DM1 and DM2. They generally occur earlier than typical age-associated cataracts seen in people without DM.
Cataracts are caused by a chemical change in the lens, which gradually goes from clear to cloudy the way the clear white of an egg becomes opaque when cooked. The exact reason why cataracts occur in DM is not known.
People with cataracts may notice their vision become blurry, hazy or dim, and that this worsens gradually over time. It often happens in both eyes, but not necessarily at the same time or at the same rate.
Read Keeping Your Focus: Eye Care, particularly the section called Other vision problems: Not common, sometimes treatable, for additional information about eye care in neuromuscular disorders.
Head, neck, and face muscle weakness
 The muscles of the neck, jaw, and parts of the head and face may weaken, especially in DM1. Facial weakness is less common and milder in DM2. Wasting of the sternocleidomastoid muscles in the neck are common in DM1 and typically absent in DM2. A "dropped head posture" is occasionally encountered.
The muscles of the neck, jaw, and parts of the head and face may weaken, especially in DM1. Facial weakness is less common and milder in DM2. Wasting of the sternocleidomastoid muscles in the neck are common in DM1 and typically absent in DM2. A "dropped head posture" is occasionally encountered.
In men, early balding in the front part of the scalp is very common, adding to the distinct appearance of DM.
Eyelids may droop (called ptosis; the “p” is silent). The chewing muscles can be affected, which makes the temples appear hollow and the face look thin.
Weak neck muscles, common in both types of DM, can make it hard to sit up quickly or lift one’s head straight up off a bed or couch. The stronger trunk muscles have to be used for these actions.
Heart difficulties
The heart can be affected in DM1 or DM2. Oddly, because DM is mostly a muscle disease, it is not the muscle part of the heart (which pumps blood) that’s most affected but rather the part that sets the rate and rhythm of the heartbeat — the heart’s conduction system.
It is common in DM1, especially after many years, to develop conduction block, which is a block in the electricity-like signal that keeps the heart beating at a safe rate. This appears to occur in DM2 as well, although there are not as many studies in this form of the disease, (rates between 20% to 37% have been reported). Arrhythmias or heart block may occasionally be very early manifestations of DM1, even when neuromuscular symptoms are mild or even unrecognized.
Fainting, near fainting, or dizzy spells are the usual symptoms of conduction block, and these should never be ignored. Such problems can be fatal.
In both forms of DM, cardiac muscle impairment also can occur, although it is not as common as conduction abnormalities.
To learn more, read Cardiac Care in DM: Lack of Symptoms May Mask Deadly Problems and Revising Cardiac Care in Muscular Dystrophies (covers different types of heart problems that occur in these disorders and how to monitor and treat them).
Insulin resistance
Fortunately, most people with DM1 and DM2 do not have diabetes, but they may develop a diabetes-like condition that is sometimes referred to as insulin resistance. This means the body makes insulin (a hormone needed for the cells to take up and use sugars), but for some reason, it takes more insulin to do the job because the muscle tissues do not respond normally to the usual amounts. High blood sugar may result from insulin resistance. The prevalence of diabetes is greater in DM2 patients than in patients diagnosed with DM1.3
Other common endocrine conditions in DM1 patients are testicular atrophy and associated low sperm count with infertility.4, 5, 6 These conditions are less common in DM2.7
Effects on internal organs
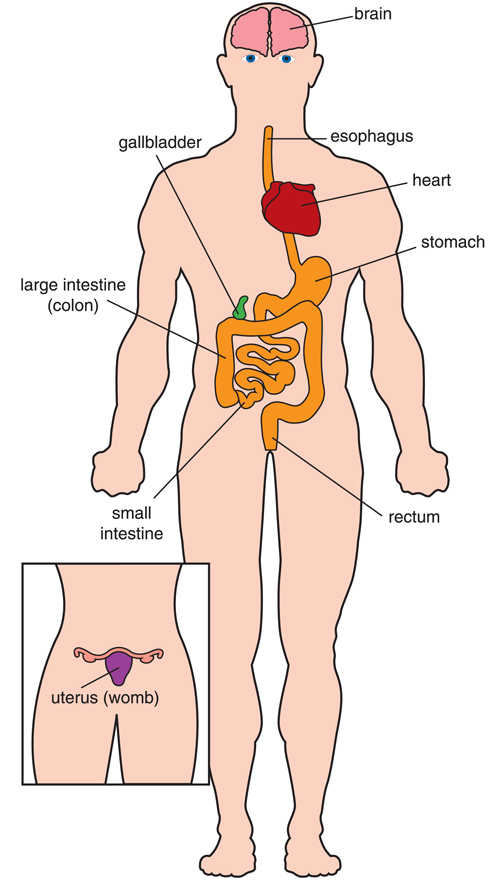 Most of the internal organs in the body are hollow tubes (such as the intestines) or sacs (such as the stomach). The walls of these tubes and sacs contain involuntary muscles that squeeze the organs and move things (food, liquids, a baby during childbirth, and so forth) through them.
Most of the internal organs in the body are hollow tubes (such as the intestines) or sacs (such as the stomach). The walls of these tubes and sacs contain involuntary muscles that squeeze the organs and move things (food, liquids, a baby during childbirth, and so forth) through them.
In DM1, many of the involuntary muscles that surround the hollow organs can weaken. These include the muscles of the digestive tract, uterus, and blood vessels. The digestive tract and uterus (womb) often are affected in type 1 myotonic dystrophy. Also, symptoms such as colicky abdominal pain, bloating, constipation, and diarrhea are common.
Abnormal action of the upper digestive tract can impair swallowing, termed “dysphagia.” Once food is swallowed, the involuntary muscles of the esophagus should take over and move food into the stomach. However, in DM1, these muscles can have spasms and weakness, causing a feeling of food getting stuck and sometimes leading to inhaling food into the lungs (aspiration), which can lead to inhalation pneumonia.
The gallbladder — a sac under the liver that squeezes bile into the intestines after meals — can weaken in DM1. People with DM probably are more likely than the general population to develop gallstones. Symptoms are difficulty digesting fatty foods and pain in the upper right part of the abdomen.
These symptoms were considered uncommon in DM2, but dysphagia of solid food, abdominal pain, and constipation have been reported by 41% to 62% of patients, a similar rate to that found in patients with DM1. Dysphagia has been proved to be relatively mild, and history of aspiration pneumonia or weight loss is rather uncommon.
Most people do not experience incontinence or urination problems in DM.
Because of weakness and uncoordinated action of the muscle wall of the uterus, women with either type of DM may experience difficulties in childbirth that can be serious for both mother and baby. These may involve excessive bleeding or ineffective labor. Preterm labor and risk of miscarriage is also more common than in women without DM. Sometimes a caesarean operation (C-section) is advised, but surgery also can be a problem in DM (see Medical Management).
Limb and hand muscle weakness
Weakness of the voluntary muscles usually is the most noticeable symptom for people with adult-onset DM. The natural history of DM1 is that of gradual progression in weakness.
The distal muscles (those farthest from the center of the body) usually are the first and sometimes the only limb muscles affected in DM1. Areas of the limbs affected may include the forearms, intrinsic muscles of the hands, and ankles. The muscles that pick up the foot when walking may weaken, allowing the foot to flop down and cause tripping and falling (foot drop). Falls and stumbles in patients with DM1 are 10 times more frequent than in a group of healthy volunteers.8 Muscles of the pelvic girdle, the hamstrings, and ankle plantar flexors are relatively spared in most cases of DM1.
In DM2, proximal muscles (closer to the center of the body) tend to show more weakness than in DM1. Weakness in the hip girdle region is often the presenting feature of DM2.9,10 Weakness in the upper part of the leg (thigh) occurs early in DM2. Weakness of thigh, hip flexor, and extensor muscles frequently impairs the ability to arise from a squat, arise from a chair, or climb stairs.7
Myotonia and muscle pain
Myotonia is a slowed relaxation following a normal muscle contraction. Myotonia is present in all patients with DM1, whereas myotonia is found in approximately 75% of patients with DM2.2, 11 Myotonia of voluntary muscles can make it hard for someone with DM1 or DM2 to relax their grip, especially in cold temperatures or under stress.3 Door handles, cups, writing by hand, and using hand tools may pose a problem, although some people never notice it. Myotonia also can affect the muscles of the tongue and jaw, causing difficulty with speech and chewing.
Myotonia can be uncomfortable and can even cause pain, although people with DM1 and DM2 also can have muscle pain that is not connected to the myotonia. Pain is more common in the legs, where myotonia cannot be demonstrated, and is one of the symptoms (along with stiffness and fatigue) that can bring patients to medical attention before the onset of symptomatic weakness. Pain in DM2 may be induced by exercise, palpation, or temperature changes.7, 12, 13 Chest pain may trigger a work-up for heart disease.
Cancer susceptibility
Myotonia is associated with higher risk of cancer. In particular, significantly elevated risk (two-fold) has been reported for cancers of the endometrium, brain, ovary, and colon. This study was based on information collected by large Swedish and Danish patient registries with more than 14,170 patients.14
References
- Turner, C. & Hilton-Jones, D. Myotonic dystrophy: Diagnosis, management and new therapies. Current Opinion in Neurology (2014). doi:10.1097/WCO.0000000000000128
- Day, J. W. et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology (2003). doi:10.1212/01.WNL.0000054481.84978.F9
- Machuca-Tzili, L., Brook, D. & Hilton-Jones, D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle and Nerve (2005). doi:10.1002/mus.20301
- Vazquez, J. A. et al. Hypothalamic-pituitary-testicular function in 70 patients with myotonic dystrophy. J. Endocrinol. Investig. Off. J. Ital. Soc. Endocrinol. (1990). doi:10.1007/BF03350681
- Ørngreen, M. C., Arlien-Søborg, P., Duno, M., Hertz, J. M. & Vissing, J. Endocrine function in 97 patients with myotonic dystrophy type 1. J. Neurol. (2012). doi:10.1007/s00415-011-6277-5
- Peric, S. et al. Hypogonadism and erectile dysfunction in myotonic dystrophy type 1. Acta Myol. (2013).
- Meola, G. & Moxley, R. T. Myotonic dystrophy type 2 and related myotonic disorders. Journal of Neurology (2004). doi:10.1007/s00415-004-0590-1
- Wiles, C. M. et al. Falls and stumbles in myotonic dystrophy. J. Neurol. Neurosurg. Psychiatry (2006). doi:10.1136/jnnp.2005.066258
- Ricker, K. et al. Proximal myotonic myopathy: A new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology (1994). doi:doi:10. 1212/WNL.44.8.1448
- Udd, B., Krahe, R., Wallgren-Pettersson, C., Falck, B. & Kalimo, H. Proximal myotonic dystrophy - A family with autosomal dominant muscular dystrophy, cataracts, hearing loss and hypogonadism: Heterogeneity of proximal myotonic syndromes? Neuromuscul. Disord. (1997). doi:10.1016/S0960-8966(97)00041-2
- Heatwole, C. et al. Patient-Reported Impact of Symptoms in Myotonic Dystrophy Type 2 (PRISM-2). Neurology (2015). doi:10.1212/WNL.0000000000002225
- Suokas, K. I., Haanpää, M., Kautiainen, H., Udd, B. & Hietaharju, A. J. Pain in patients with myotonic dystrophy type 2: A postal survey in Finland. Muscle and Nerve (2012). doi:10.1002/mus.22249
- George, A., Schneider-Gold, C., Zier, S., Reiners, K. & Sommer, C. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch. Neurol. (2004). doi:10.1001/archneur.61.12.1938
- Gadalla, S. M. et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA - J. Am. Med. Assoc. (2011). doi:10.1001/jama.2011.1796
- Meola, G. et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul. Disord. (2003). doi:10.1016/S0960-8966(03)00137-8
- Udd, B. & Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. The Lancet Neurology (2012). doi:10.1016/S1474-4422(12)70204-1
- Ekström, A. B., Hakenäs-Plate, louise, Tulinius, M. & Wentz, E. Cognition and adaptive skills in myotonic dystrophy type 1: A study of 55 individuals with congenital and childhood forms. Dev. Med. Child Neurol. (2009). doi:10.1111/j.1469-8749.2009.03300.x
- Douniol, M. et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child Neurol. (2012). doi:10.1111/j.1469-8749.2012.04379.x
- Angeard, N. et al. A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromuscul. Disord. (2011). doi:10.1016/j.nmd.2011.04.009
- Echenne, B. et al. Myotonic dystrophy type I in childhood. Long-term evolution in patients surviving the neonatal period. Eur. J. Paediatr. Neurol. (2008). doi:10.1016/j.ejpn.2007.07.014
- Bhakta, D., Lowe, M. R. & Groh, W. J. Prevalence of structural cardiac abnormalities in patients with myotonic dystrophy type I. Am. Heart J. (2004). doi:10.1016/j.ahj.2003.08.008
- Bassez, G. et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology (2004). doi:10.1212/01.WNL.0000144343.91136.CF
- Wesström, G., Bensch, J. & Schollin, J. Congenital myotonic dystrophy. Incidence, clinical aspects and early prognosis. Acta Paediatr. Scand. (1986).
- Campbell, C., Levin, S., Siu, V. M., Venance, S. & Jacob, P. Congenital myotonic dystrophy: Canadian population-based surveillance study. J. Pediatr. (2013). doi:10.1016/j.jpeds.2012.12.070
- Harley, H. G. et al. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum. Genet. (1993).
- Whelan, D. T., Carson, N. & Zeesman, S. Paternal transmission of the congenital form of myotonic dystrophy type 1: A new case and review of the literature. Am. J. Med. Genet. (2002). doi:10.1002/ajmg.10141
- Darras, B. T. & Volpe, J. J. Muscle Involvement and Restricted Disorders. in Volpe’s Neurology of the Newborn (2017). doi:10.1016/B978-0-323-42876-7.00033-8
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Search for Clinical Trials
Learn More -

Grants at a Glance
Read More

