
Debatable Destinies

Duchenne muscular dystrophy carriers carry on, despite uncertainty
When Rena Szymanski turned 40, she expected to slow down a bit. She never had been athletic, and it sometimes seemed to her that climbing stairs was harder for her than it was for other people, but she thought her strength was “normal” in general.
Soon after her birthday, though, she noticed increasing weakness. “I didn’t know what was wrong with me,” says Szymanski, who until recently was working at a stressful job an hour away from her home in Stony Point, N.Y. To her knowledge, there is no history of muscle disease in her family.
After first consulting a rheumatologist (inflammatory disease specialist), who didn’t find anything, she traveled to Columbia University Medical Center in New York and underwent a muscle biopsy and DNA testing.
The diagnosis was “manifesting carrier of Duchenne muscular dystrophy,” with a mutation in the gene for the muscle protein dystrophin.
The classic definition of a disease carrier is someone who can pass the disease to others but doesn’t show symptoms. But manifesting carriers are carriers who show (manifest) symptoms of the disease themselves. Some estimates are that as many as one in 10 carriers of DMD or its less severe variant, Becker MD, have signs of the disease themselves. Usually, the signs are relatively mild — but not always.
“Just to take my daughter shopping at a mall is a big, exhausting thing,” says Szymanski, who is considering getting a power chair for long distances. So far she still can walk short distances and drive, but she wonders what the future holds. Unfortunately, no one seems to have any clear answers.
There was something wrong
Like Rena Szymanski, Karen Smith of Madison, Ala., knows of no family history of muscular dystrophy. So when their infant daughter, Chloe, wasn’t meeting the usual motor skills milestones, the former elementary school teacher and her husband, a physicist, were taken by surprise.
Karen’s mother, a nurse, suspected something was wrong, but the family’s pediatrician recommended waiting until Chloe was 6 to 9 months old before getting concerned.
“At her 9-month checkup, they gave us a questionnaire,” Karen recalls, “with questions like, ‘Does the baby crawl? Does she pull herself into a sitting position?’ I couldn’t answer yes to any of them.”
A referral to a cerebral palsy clinic revealed that Chloe was “significantly delayed.”
“I sat in the parking lot and cried,” Karen says. “I felt so overwhelmed. I was thinking, I don’t want to do this.”
In the chair a lot more
 The Smiths found their way to Jean Teasley, a neurologist in nearby Huntsville who specializes in neuromuscular disease, and embarked on blood tests, MRI scans and brain-wave studies. These revealed nothing out of the ordinary except elevated serum levels of creatine kinase (CK), an enzyme that leaks out of damaged muscle cells.
The Smiths found their way to Jean Teasley, a neurologist in nearby Huntsville who specializes in neuromuscular disease, and embarked on blood tests, MRI scans and brain-wave studies. These revealed nothing out of the ordinary except elevated serum levels of creatine kinase (CK), an enzyme that leaks out of damaged muscle cells.
More tests, including a muscle biopsy and DNA analysis, followed. Then, at the end of 2001, with Chloe just over a year old, the Smiths had their answer.
 “Dr. Teasley called us the week before Christmas and said Chloe had been diagnosed as a manifesting carrier of Duchenne muscular dystrophy,” Karen recalls. Her muscle biopsy sample showed a mosaic pattern of dystrophin staining (meaning some fibers had dystrophin and others didn’t), and a deletion in one of her two dystrophin genes.
“Dr. Teasley called us the week before Christmas and said Chloe had been diagnosed as a manifesting carrier of Duchenne muscular dystrophy,” Karen recalls. Her muscle biopsy sample showed a mosaic pattern of dystrophin staining (meaning some fibers had dystrophin and others didn’t), and a deletion in one of her two dystrophin genes.
Today, 7-year-old Chloe can do most of what she wants to do, her mother says, but not quickly and not for long. “Fatigue is a gigantic issue,” Karen says, as are muscle spasms in her legs.
When Chloe grew too big for strollers and shopping carts, her parents acquired a power chair for distances. It’s now used in the house as well. “In the last six months or so, we’ve seen her in her chair a lot more,” Karen says, describing it as a “hard transition for me” but “an amazing transformation” for Chloe.
“I realized that, for the first time when we were out, she was very independent. Before the power chair, I never had to say ‘Chloe, come here,’ or ‘Chloe, don’t touch that.’”
The Smiths got involved with local MDA activities, and last year Chloe served as goodwill ambassador for Alabama.
Where do we end up?
The Smiths have met people through MDA support groups and learned more about manifesting carriers of DMD, but no one seems to be in their situation.
“We’ve gotten some information about adults who have been diagnosed as manifesting carriers,” Karen says, “and they have fatigue and heart problems and a range of minor effects compared to boys with Duchenne dystrophy. I’ve asked Dr. Teasley, ‘Where do we end up?’ She said, ‘I can’t tell you right now, but as we follow Chloe, we’ll begin to know.’”
The X files
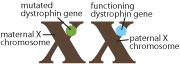
In 1961, British geneticist Mary Lyon proposed the existence of the phenomenon known as X-chromosome inactivation, a mechanism that would make the “dosage” of X-chromosome genes equal for males and females. Males have one X chromosome, which they get from their mothers, and a Y chromosome from their fathers. Females have two X chromosomes, one from each parent.
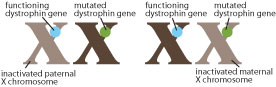
Lyon was right; in fact, the process of X-chromosome inactivation also is called “lyonization.” Early in the embryonic development of a female, either the X chromosome from the mother (maternal X) or the one from the father (paternal X) is inactivated. The choice of which chromosome to inactivate is random. In each cell, there’s a 50 percent chance that either the maternal or paternal X chromosome will be inactivated, with the other left active.
But if you flip a coin 100 times, you won’t always get 50 heads and 50 tails. Sometimes you might get 40 heads and 60 tails, or even 30 heads and 70 tails. The same holds true when the maternal or paternal X chromosome is randomly inactivated 100 times: You may end up with 50 maternal and 50 paternal X-chromosome inactivations, or with other combinations, such as 60 maternal and 40 paternal inactivations.
Most of the time, it doesn’t matter how many inactivated maternal and paternal X chromosomes a female has. But when there’s a mutation in an X-chromosome gene, such as in the gene for dystrophin, it matters a lot.
If a girl or woman has to rely on too many X chromosomes with the dystrophin gene mutation (meaning the Xs with the functional dystrophin genes are mostly inactivated), she’s likely to develop symptoms of DMD or BMD.
In very rare instances, a girl may lack a second X chromosome entirely, or her second X may have sustained serious damage. In these cases, she makes little or no dystrophin (depending on the type of dystrophin mutation), and she develops DMD or BMD just as a boy would.
Distribution matters
 MDA research grantee George Karpati, a professor of neurology at McGill University in Montreal, where he also directs a neuromuscular research laboratory, has studied X inactivation in DMD carriers since the early 1990s.
MDA research grantee George Karpati, a professor of neurology at McGill University in Montreal, where he also directs a neuromuscular research laboratory, has studied X inactivation in DMD carriers since the early 1990s.
“Most carriers are in the 50-50 to 60-40 range,” Karpati says, referring to the percentage of maternal and paternal X chromosomes that are inactivated. “In manifesting carriers, it’s often still 60-40, but those are the ones who also have a distribution problem. It’s not just the absolute amount of dystrophin that counts, but getting a good, even distribution [in muscle fibers].”
 The percentage of maternal and paternal X chromosomes that have been inactivated is generally determined from a blood sample. A muscle biopsy is required to estimate the amount of dystrophin protein being produced and to see its location in muscle fibers.
The percentage of maternal and paternal X chromosomes that have been inactivated is generally determined from a blood sample. A muscle biopsy is required to estimate the amount of dystrophin protein being produced and to see its location in muscle fibers.
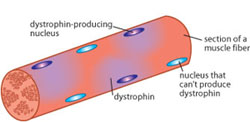
Mature muscle fibers have hundreds to thousands of cell nuclei. Each nucleus was originally part of a small, immature cell, and in females, each had an equal chance of having its maternal or paternal X chromosome inactivated early in development.
In a female DMD carrier, mature fibers have some nuclei that produce dystrophin and others that don’t.
 “Since those nuclei are very close to each other, if half or so don’t produce dystrophin, the other half almost cover their territory,” Karpati says. “But sometimes distribution is not even. If you have a centimeter of muscle fiber that contains all mutant dystrophin genes, that segment is doomed. There are segments of muscle fibers here and there where there’s no dystrophin, and they break down. Eventually their regenerative capacity declines, just like in Duchenne dystrophy.”
“Since those nuclei are very close to each other, if half or so don’t produce dystrophin, the other half almost cover their territory,” Karpati says. “But sometimes distribution is not even. If you have a centimeter of muscle fiber that contains all mutant dystrophin genes, that segment is doomed. There are segments of muscle fibers here and there where there’s no dystrophin, and they break down. Eventually their regenerative capacity declines, just like in Duchenne dystrophy.”
And sometimes X-chromosome inactivation is so far from 50-50 maternal versus paternal chromosomes that the distribution is said to be “skewed” in one direction or the other. A skewing of inactivation toward the dystrophin-competent X chromosome leads to a manifesting carrier. To get symptoms nearly as severe as those in a boy with DMD, he says, “you have to have a badly skewed situation.”
Karpati advises parents of girls with symptoms as severe as Chloe Smith’s that “this is basically a Duchenne dystrophy,” resulting from the same mechanisms that operate in boys with the disease. However, a girl’s entire musculature isn’t as likely to be affected as a boy’s, because of spotty dystrophin production here and there, thanks to the girl’s second X chromosome.
Heart troubles
Karpati estimates that some 10 percent to even 20 percent of DMD carriers could be called manifesting “if we looked with a fine-tooth comb and sophisticated measurements.” But he estimates that only 3 percent to 5 percent of carriers manifest noticeable weakness.
“A 60-40 skew is usually not a problem,” Karpati says, especially if the distribution of dystrophin is relatively even. But a skew of 75-25 in favor of inactivation of dystrophin-competent X chromosomes produces noticeable symptoms, particularly if the dystrophin distribution is uneven.
Some female carriers of DMD have cardiomyopathy (cardiac muscle degeneration) but manifest few or no other symptoms. That’s because heart muscle is different from other muscles in the body, Karpati explains.
Cardiac muscle cells don’t have the multinuclear fiber structure that skeletal muscle cells do, which leaves them more vulnerable to the effects of X chromosomes that can’t make dystrophin. They also lack the repair mechanisms that skeletal muscle fibers have.
However, Karpati notes, cadiac muscle cells don’t have the same dependency on dystrophin as skeletal fibers do, so they can function with less of the protein.
Time and chance
“Time can be a friend of the carrier,” says MDA grantee Eric Hoffman, who directs the Research Center for Genetic Medicine at Children’s National Medical Center in Washington.
Early in the life of a female embryo, he explains, there’s a random mixing of cells that have inactivated the maternal or paternal X chromosome.
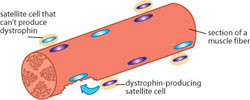
Some of these cells will go on to become part of a long muscle fiber. If there are enough dystrophin-producing nuclei and if they’re fairly evenly distributed, the fiber will undergo what Hoffman calls “biochemical normalization,” meaning their dystrophin production is adequate to preserve the fiber.
Other cells will become a type of muscle stem cell called a satellite cell, to be called in for repair work when a fiber is in trouble. If a degenerating fiber is lucky enough to be repaired by cells that can produce dystrophin, it’s likely to survive. If it’s repaired by cells that can’t make dystrophin, it’s likely to die.
Over time, the survivors come to predominate, a process Hoffman calls “genetic normalization.” However, the dystrophin-producing fibers may be surrounded by the remains of dead fibers that have become scar tissue and cause permanent weakness, so this can be somewhat of a hollow victory.
“You have this constant selection against dystrophin-negative fibers,” Hoffman says. “[Fibers] either become dystrophic and die, or they’ll have a chance to be regenerated from dystrophin-positive stem cells. Because that selection process is constant throughout a lifetime, you’re always losing dystrophin-negative fibers, with the option of growing back positive fibers.”
Unfortunately, that option doesn’t seem to last forever. As a female ages, she may accumulate many dystrophin-negative fibers that are beyond rescue, and she may run low on dystrophin-positive repair cells.
For manifesting carriers, Hoffman says, “There’s a race between the genetic and biochemical processes that help the patient, and the development of fatty, dystrophic muscle and failed regeneration. If the muscle can do OK for long enough, these normalization processes can almost fix it. But if the race is lost to scar tissue formation, then you have Duchenne dystrophy.”
Accepting and moving on
“I’m not able to do the things that I really want to do,” Rena Szymanski says. In the past year, the muscles in her hips have deteriorated a little more. Sometimes she uses a cane, and she hopes her insurance will cover a wheelchair.
“I recently registered with MDA, which is a good thing,” she says. Her heart function is good, and she’s able to get more rest since leaving her job. She’d like to know more people in her situation. “I’d like to feel like I’m not the only one. I’d like to feel that somebody has in common the same things that I feel.”
 Chloe Smith has recently learned to read, and she likes to play quietly with her friends. “They sit on the floor and color or do art,” Karen says. “She likes activities where she doesn’t have to move around. She’ll go outside, stay five minutes, and she’s ready to come in.” Her muscle spasms have improved with the muscle relaxant baclofen.
Chloe Smith has recently learned to read, and she likes to play quietly with her friends. “They sit on the floor and color or do art,” Karen says. “She likes activities where she doesn’t have to move around. She’ll go outside, stay five minutes, and she’s ready to come in.” Her muscle spasms have improved with the muscle relaxant baclofen.
Recently, the Smiths tried giving Chloe prednisone, an anti-inflammatory drug that has shown benefits in prolonging function in boys with DMD, but the side effects were intolerable, and they stopped the drug.
Chloe falls a lot, sometimes very hard. “In the last year, we’ve had some instances where she has gotten down and couldn’t get up. She got on the floor of the car one day and couldn’t get up.”
Karen says they’ve recently built a new house, one that can accommodate a wheelchair, now that they have “accepted it’s going to be a part of her life.”
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
