
SMA Research
 This decade, for the first time, scientific research in spinal muscular atrophy (SMA) has yielded enough information to allow the pursuit of several avenues of disease treatment and prevention.
This decade, for the first time, scientific research in spinal muscular atrophy (SMA) has yielded enough information to allow the pursuit of several avenues of disease treatment and prevention.
Most are centered around increasing cellular levels of a protein called SMN (survival of motor neuron), first identified in the mid-1990s as the root cause of almost all cases of SMA.
A few scientists are studying exactly what SMN does to see if its absence might be compensated for via some other gene or protein.
The curious case of a backup gene
During the 1990s, investigators, many of whom had MDA support, uncovered a fundamental clue about SMA that would have profound implications for its treatment: While everyone with SMA lacked or had mutations in genes known as SMN1, located on chromosome 5, everyone with the disease also had at least one functional copy of a gene called SMN2 located nearby.
Researchers soon learned the difference between SMN1 and SMN2: Most of the protein produced from the DNA instructions carried by the SMN1 gene is so-called “full-length” SMN, a protein whose functions are incompletely understood but apparently necessary to the health of motor neurons. By contrast, most of the protein produced from neighboring SMN2 genes is a short form of SMN, which is not functional.
The key word in that sentence is “most.” About 20 percent of the protein molecules produced from the SMN2 gene are in fact full-length, fully functional SMN. And the more SMN2 activity there is, the better an organism tolerates the loss of SMN1.
Researchers quickly learned that the number of SMN2 “backup gene” copies varies in the population in general. In people with SMA, who lack functional SMN1 genes, the more SMN2 copies someone has, the better he or she is likely to fare.

Babies born with no SMN1 genes and one or two SMN2 genes usually have the very severe form of the disease known as type 1 SMA, in which severe weakness and respiratory muscle impairment usually lead to early death.Type 2 SMA is less severe and allows for a longer life span, albeit with disabilities, and people with this form of the disease generally have about three copies of SMN2.
Type 2 SMA is less severe and allows for a longer life span, albeit with disabilities, and people with this form of the disease generally have about three copies of SMN2.
The least severe SMA cases, known as type 3 (with some physicians adding a type 4) generally occur when there are at least three SMN2 genes present.
The correlation between number of SMN2 genes and severity of SMA, however, is only approximate. Research to identify other factors is under way.
Just what SMN does in nerve or other cells, and whether its presence is required during development of the nervous system to prevent permanent damage, remain incompletely answered questions.
Augmenting SMN by modifying SMN2 gene instructions
For Adrian Krainer at Cold Spring Harbor Laboratory on New York’s Long Island, the preferred strategy to find a cure for SMA involves changing the SMN2 gene’s instructions by altering a process called splicing, a kind of molecular cutting and pasting that cells perform.
“Changing the splicing that underlies the defect has a certain elegance,” says Krainer, who has MDA funding for this work and is collaborating with Isis, a drug development company in Carlsbad, Calif. However, he says, “there are different ways of solving the SMA problem, many approaches to pursue in parallel.”
Since 1983, Krainer has been working on RNA splicing, a method cells use to refine a genetic message as it moves from its permanent form as DNA to a “rough draft” RNA molecule and then to one or more “final” RNA versions, known as messenger RNAs, that provide direct instructions for the cell to make protein molecules.
One way to change how a cell splices the RNA on its way to becoming a final copy is by hiding parts of the RNA strand from the cell’s processing mechanism, thereby changing the final structure of the messenger RNA. Krainer and others accomplish this sleight of hand by using synthetic material called “antisense,” which specifically targets, sticks to and prevents a cell from “seeing” an RNA section.
Without treatment, the section of the SMN2 gene called exon 6 is spliced directly to the section called exon 8, and exon 7 is left out of the final RNA. But using antisense to block a “clip out exon 7” signal on the RNA coaxes the cell to include this critical exon and ultimately make full-length SMN protein.
Krainer is now testing the various antisense molecules in two strains of laboratory mice with a disease closely resembling human SMA.
Before proceeding toward human testing, which Krainer hopes will be within the next few years, he and his colleagues want to find out in the laboratory how important the timing of SMN treatment is and determine whether an increase in SMN after the onset of SMA can stop disease progression, or even reverse the disease process.
Christian Lorson, an MDA grantee at the University of Missouri-Columbia, is also pursuing strategies to get more full-length SMN out of the SMN2 gene, although that’s not the only approach to SMA that intrigues him.
Lorson says he’s working on several experimental approaches to treating SMA. Two of them, both aimed at changing splicing, are known as “trans-splicing” and “bifunctional” RNAs.
Rather than just masking part of the SMN2 RNA with antisense and hoping the cell will include exon 7, Lorson says he prefers to take a firmer approach to “persuading” the cell to include exon 7 by providing that piece of RNA along with blocking the original instructions.
He and graduate student Tristan Coady developed an antisense molecule that overlaps the place where exon 6 would be spliced to exon 8, preventing that from happening. The cell then has to “choose” between no splicing at all and splicing in the new exon 7 provided by the scientists, Lorson says. “When faced with that choice,” Lorson says, the cell considers splicing in the newly supplied exon 7 RNA (“trans-splicing”) the better option.Lorson’s lab group is also working on a variation of this strategy, using what he calls “bifunctional” RNAs. A bifunctional RNA molecule is a combination of a piece of antisense that targets the SMN2 gene at the point where exon 7 normally would be spliced out, tethered to another piece of RNA that specifically attracts cellular proteins normally involved in splicing that encourage exon 7 inclusion.
Lorson’s lab group is also working on a variation of this strategy, using what he calls “bifunctional” RNAs. A bifunctional RNA molecule is a combination of a piece of antisense that targets the SMN2 gene at the point where exon 7 normally would be spliced out, 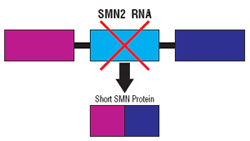 tethered to another piece of RNA that specifically attracts cellular proteins normally involved in splicing that encourage exon 7 inclusion.
tethered to another piece of RNA that specifically attracts cellular proteins normally involved in splicing that encourage exon 7 inclusion.
Stabilizing short SMN
Lorson, who has fingers in almost every part of the SMA research pie, also is working on a unique strategy that he says isn’t widely endorsed but is gaining adherents: improving the stability of the short protein made from the SMN2 gene.
In collaboration with Cheng-Wei Tom Chang, a chemist at Utah State University in Logan, Lorson and Virginia Mattis, another graduate student, are experimenting with antibiotics known as aminoglycosides, which cause cells to ignore, or “read through,” stop signals in RNA and make longer protein molecules.
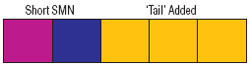 The “read-through” product made from the SMN2 gene still lacks exon 7, but it has a longer tail after exon 7 than it normally would have.
The “read-through” product made from the SMN2 gene still lacks exon 7, but it has a longer tail after exon 7 than it normally would have.
“It may be that the read-through protein is not by itself intrinsically more functional,” Lorson says, “but in combination with full-length SMN, it enhances and stabilizes the normally low SMN levels. At the end of the day, we want more SMN protein, regardless of mechanism. That would be great.”
His hypothesis, now being studied, is that the read-through SMN protein forms a two-part molecule with whatever full-length SMN is available, which improves its stability and function. Normally, he says, full-length SMN molecules associate with each other and form this type of two-part structure, but short SMN that lacks both exon 7 and a longer tail can’t participate in this interaction.
He says that if the benefit they’re seeing in cells “is coming about because of full-length SMN” rather than directly from short SMN, “that’s fine.”
The next step will be to see if it helps SMN-deficient mice.
Raising SMN levels by increasing total output from the SMN2 gene
Because some percentage of output from the SMN2 gene is full-length SMN protein, it stands to reason that increasing the total output from the gene would raise levels of both compounds.
One way to increase the activity of a gene is with compounds known as histone deacetylase inhibitors, or HDAC inhibitors, which keep DNA messages open for transcription into RNA and ultimately cause more protein molecules to be made. Several ongoing drug trials in people with SMA are making use of this phenomenon.
Sodium phenylbutyrate and valproic acid are in this category and are being tested in patients. The Boston pharmaceutical company Paratek is investigating whether antibiotics in the tetracycline family have HDAC inhibitory activity and can increase SMN production. According to a 2008 press release, the company has been encouraged by early evidence.
Elliot Androphy, who has MDA supportfor SMA research at the University of Massachusetts-Worcester, believes HDAC inhibitors “hold promise” but that they also come with some caveats. “It’s an idea with potential and some experiments suggest these may be of benefit,” he says. “But we really don’t know how they may be working.” Androphy and others are concerned about the lack of specificity of HDAC inhibitors, which are likely to increase output from a number of genes, not just the SMA target gene.
Transferring genes or stem cells
Another strategy for raising full-length SMN levels is to deliver the compound via genes that make it, using stem cells or other delivery vehicles.
California Stem Cell in Irvine is developing cell transplant therapies and has named SMA and ALS (amyotrophic lateral sclerosis) as its first targets. According to Chief Operating Officer Chris Airriess, the company hopes to test stem cells in type 1 SMA patients later this year.
At the U.S. Food and Drug Administration, Jakob Reiser has MDA support to study the delivery of the SMN1 gene via gene therapy. Reiser, a molecular biologist who specializes in developing viral gene delivery vehicles, is working closely with neurosurgeon Nicholas Boulis at Emory University in Atlanta.
Reiser says they’ll inject the SMN1 gene at various time points into mice that have an SMA-like disease that allows them to survive for six to seven months with a fair percentage of their motor neurons intact. The investigators will deliver the gene, encased in a viral transporter, into mouse muscles, with the expectation that it will be transported from muscle fibers to motor neurons via connecting nerve fibers.
The viral transporter carrying the SMN1 gene is an altered lentivirus, which has a good reputation for reaching the nervous system. Reiser and Boulis also plan to use a molecular “on switch” (“promoter”) called Hb9, which should cause the gene to be activated and the SMN1 protein produced in motor neurons and probably not in other types of cells.
Either gene therapy or stem cells also could be used to deliver other therapeutic agents, such as “neurotrophic” (nerve-nourishing) proteins. Reiser and Boulis plan to study the effects of transferring the gene for one such protein, called insulin-like growth factor 1 (IGF1), alone and in combination with the SMN1 gene, in their SMA mice.
In addition, stem cells are expected to provide a valuable research tool. In December 2008, Lorson was part of a scientific team coordinated by Clive Svendsen at the University of Wisconsin-Madison, that showed SMA-affected motor neurons can be developed in the laboratory from skin cells taken from an SMA patient. These, he says, will provide a valuable environment in which to “ask biological questions” about the SMA disease process as well as develop therapeutics in a human disease context.
Using stem cells as therapeutic agents themselves, Lorson says, is not as simple as it’s sometimes depicted, although truly exciting breakthroughs seem to be developing at an unprecedented rate. “There are biological barriers designed to keep the central nervous system protected and isolated,” he notes, and these would have to be breached for new motor neurons to grow and activate muscle fibers at significant distances from the spinal cord.
Beyond boosting SMN
Umrao Monani, who has received MDA funding for SMA studies at Columbia University’s Motor Neuron Center in New York, wants to know more about what SMN does and when it does it. “I am convinced that SMN is multifunctional,” he says, “and that the level of SMN you have may determine how many functions of SMN you can maintain.”
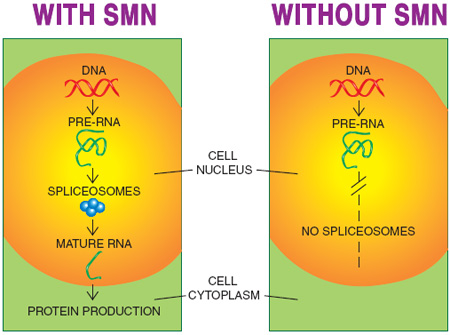
One role of full-length SMN that’s universally agreed upon is its participation in building the so-called spliceosomes in the cell nucleus. Spliceosomes are the structures where RNA splicing, in which draft RNA becomes messenger RNA, is conducted.
With so much emphasis on changing the splicing of SMN2 RNA so that full-length SMN is made, it’s confusing to think of SMN itself as part of the splicing machinery that affects the manufacturing of various proteins, but that’s a crucial concept. So far, it’s not clear which RNAs are most affected by a lack of SMN and therefore deficient spliceosome activity, but it’s certain that some are. If any of those affected are needed during development of the nervous system, their absence early in life could take a heavy toll.
Monani is among those who believe SMN’s role is not limited to spliceosome formation. He’s particularly interested in SMN’s potential involvement in transporting RNA molecules down the long fibers (axons) from motor neurons in the spinal cord to muscles, especially during the development of the axons. “One scenario is that SMN transports a particular molecule to the neuromuscular junction [where nerve fibers and muscle fibers intersect],” Monani says.
He and his colleagues published a paper last summer supporting this idea by showing that mice with insufficient functional SMN early in life don’t form normal neuromuscular junctions and have impaired transmission of signals from nerve to muscle. Monani and colleagues, however, draw a positive conclusion from this otherwise concerning finding, suggesting that maintaining function at the neuromuscular connection might be a promising treatment avenue for SMA.
Livio Pellizzoni, who’s also part of Columbia’s Motor Neuron Center, believes it’s important to understand more about SMN function and the time period during which the protein is essential before relying on SMN-based therapies as treatments for SMA.
Clearly, he says, SMA can be prevented by increasing SMN levels before birth, but it’s unclear whether it can be effectively treated or cured by increasing them later on.
He cites a landmark MDA-supported study published in 2000 that shows positive outcomes for mice bred so as not to have any copies of the mouse SMN genes and eight copies of the human SMN2 gene. Although the researchers achieved full rescue of the mice by increasing SMN protein levels, Pellizzoni notes that they only prevented the disease; they didn’t reverse it.
Likewise, humans lacking SMN1 genes benefit from having multiple SMN2 genes. But, like the mice, they have these genes from birth; they don’t acquire them later on.
Pellizzoni, who had MDA funding from 2004 to 2006 to study SMN in mice, says the protein appears to have many roles in nerve cells, most of which are incompletely understood. Studying all of SMN’s functions “gives you an opportunity to understand the mechanisms of this disease, so that you have additional ways to identify targets and develop therapies.”
While he’s in favor of pursuing strategies to change SMN2 gene splicing or increase its total output, he says identification of additional targets alsoneeds to be pursued. “We need to see if there’s a gene X that might influence the SMA phenotype [clinical picture] and be amenable as a drug target.” And, he says, “It would be great to have an animal model showing the time window for intervention.”
Newborn screening would allow earlier treatment
If it becomes clear that very early treatment, either with SMN augmentation strategies or other measures, can improve survival and quality of life for people with SMA, the case for newborn screening to detect the disease becomes a strong one.
“We ought to consider newborn screening,” says Tom Prior, who directs the Molecular Pathology Laboratory at Ohio State University in Columbus. Prior, who received MDA funding from 1996 to 2001 to develop diagnostic testing for SMA, advocates screening newborns for SMA so that babies can be enrolled in a clinical trial or receive whatever treatments are developed as early as possible. He also advocates screening for carriers of the disease so that parents can make informed reproductive decisions.
Carrier and prenatal testing can decrease disease incidence
“We have to hit SMA at both ends,” Prior says. “We need newborn screening for the betterment of the child and the ability to enroll in clinical trials immediately, and we need carrier testing to decrease the incidence of the disease.” (Carrier testing is available through several clinical laboratories, including the one at Ohio State.)
In an article in the November 2008 issue of Genetics in Medicine, Prior recommends that SMA carrier testing be offered to all couples; that formal genetic counseling services be made available to anyone requesting this testing; that all identified carriers be referred for follow-up genetic counseling; and that parents be informed that negative carrier test results reduce but don’t completely eliminate the possibility of having an affected child, and that carrier testing can’t predict disease severity.
“As is true for all carrier screening programs,” notes Prior, representing the Professional Practice and Guidelines Committee of the American College of Medical Genetics, “the testing [should be] voluntary. Informed consent and the usual caveats must be addressed, including assurance of confidentiality, paternity issues, discrimination, self-esteem and cost.”
Prenatal testing to identify SMAaffected fetuses and preimplantation genetic diagnosis to identify SMAaffected embryos also are available at some centers. (For more about these options, see “Do You Really Want to Know?” in the July-August 2007 Quest.)
Non-chromosome 5 SMA also being studied
In 2008, Lisa Baumbach-Reardon at the University of Miami (Fla.) and colleagues announced the culmination of their decade-long search for the cause of a severe, infantile-onset form of SMA that isn’t caused by SMN deficiency but arises from a mutation in an X-chromosome gene.
Baumbach-Reardon, who received three MDA grants for this search between 1999 and 2005, led a study team with Alfons Meindl at Technical University Munich (Germany) that identified this gene last year.
The gene, known as UBE1, carries instructions for ubiquitin-activating enzyme 1, which normally helps attach a molecular tag to proteins to mark themfor destruction by a cell. Altered function of this protein disposal system is the likely mechanism by which X-linked SMA occurs. In addition to helping the small number of families affected by this rare form of SMA, understanding how UBE1 defects cause the disease may lead to further insights about motor neurons that will prove useful with respect to chromosome-5 SMA as well.
Baumbach-Reardon now has MDA support to extend her X-linked SMA studies.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
