
New Directions in Autoimmune Disorders

Can an immune response be rerouted to treat disease?
What do inflammatory muscle diseases (myositis), myasthenia gravis, Lambert-Eaton syndrome, and dozens of other disorders like lupus and rheumatoid arthritis, have in common?
In all these and more, the body’s immune system, which normally detects and destroys invading microbes that cause illness, somehow has taken a wrong turn, and is attacking its own tissues, causing what’s known as an “autoimmune” (self-immune) disease.
In myositis, the target is mostly muscle fibers. In myasthenia gravis, it’s the parts of muscle cells that normally receive signals from nerve cells (the acetylcholine receptors), and in Lambert-Eaton syndrome, it’s the parts of nerve cells that signal muscle cells.
But it’s not only the targets of the immune system that differ among the various autoimmune disorders. These disorders also differ with respect to which part of the immune system predominates in causing the problem and what kind of solution might work best for solving it.
Highway patrol
The first step in any immune response is the perception of a threat (real or not) by specialized cells called macrophages (“big eaters”) and dendritic (branching) cells.
These cells patrol the body’s highways and can approach an invader, usually a microbe, such as a bacterium, and engulf it. They then digest its proteins and display pieces of them on the cell surface, like red flags to alert the immune system to the possible danger.
Each red flag is called an “antigen” and can generate an attack against itself and any other molecule that looks like it. The cells that show it to the immune system, such as macrophages and dendritic cells, are called “antigen-presenting” cells.
In autoimmune disease, the body’s own proteins become antigens.
Response enforcement
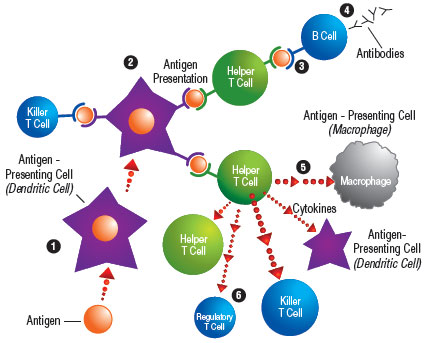 Antigen-presenting cells draw the attention of T (thymus) cells, the chief effectors (producers) of an immune response. They originate in an organ in the chest called the thymus and then migrate to the lymph nodes and elsewhere via the bloodstream.
Antigen-presenting cells draw the attention of T (thymus) cells, the chief effectors (producers) of an immune response. They originate in an organ in the chest called the thymus and then migrate to the lymph nodes and elsewhere via the bloodstream.
Two types of effector T cells can be activated by antigens: killer T cells and helper T cells. Killer T cells kill their targets directly. For instance, they make contact with virus-infected cells and destroy them by making holes in their membranes.
Helper T cells, on the other hand, attack indirectly, by telling other cells to do the killing. After making contact with an antigen-presenting cell, they secrete compounds called cytokines, which can travel relatively long distances and enhance the power of killer T cells and macrophages.
They also can make direct contact with B (bone marrow) cells, immune-system cells that originate in the bone marrow and travel via the circulation. The helper T cells pass along the message they received from the antigen-presenting cells and tell B cells to make molecular bullets called antibodies against specific antigens.
Antibodies are proteins that home in on antigens and either mark them for destruction by other components of the immune system or neutralize them directly.
Autoimmunity can be mostly cell-based, with dendritic cells, macrophages and killer T cells at center stage; or primarily antibodybased, with B cells and antibodies doing most of the destruction.
Dermatomyositis (DM) and polymyositis (PM) are thought to be mostly cell-based autoimmune diseases, while the destruction in myasthenia gravis (MG) is carried out mostly by antibodies.
Restoring peace
All immune responses, even appropriate ones, take a toll on the body. Cells harboring viruses die, sometimes impairing the work of the organs where they reside, and cytokines can cause tissues to become painfully inflamed.
Autoimmune responses are even more damaging than normal immune responses, because the immune system’s “friendly fire” kills large numbers of cells the body needs as it tries to stamp out an illusory enemy invader.
In any immune response, there’s a need for the immune system to declare victory (or occasionally defeat) and withdraw. In fact, another kind of T cell, called a regulatory T cell, has recently been discovered to be involved in the process of turning off an immune response and restoring peace in the body.
In autoimmune conditions, not only are the antigens derived from the body’s own proteins; but to make matters worse, the immune system doesn’t seem to know when to quit. The resulting prolonged engagements manifest themselves as chronic diseases, often requiring years of medications that suppress the immune system.
Prednisone, azathioprine, methotrexate and cyclophosphamide are some of the older drugs used to suppress the immune system. They can be very effective, but they have widespread effects, not all of them desirable.
Mycophenolate mofetil, rituximab, infliximab and etanercept are newer agents, targeted to specific parts of the immune system. But even these newer drugs have side effects, particularly reduction of the body’s ability to fight infection.
Although understanding of autoimmunity and treatments for it has come a long way in the last 20 years, there’s more work to be done, and MDA-supported researchers are developing some new ideas.
Interfering with interferons
Until the 1970s, antigen presenters known as dendritic cells didn’t get much attention. Now, however, they’re believed to play a major role in the immune system.
The first dendritic cells to be identified are known as “myeloid” (from the bone marrow). Myeloid dendritic cells pick up suspicious materials and bring them to lymph nodes, where B and T cells decide whether they warrant an immune-system attack or not, says MDA grantee Steven Greenberg at Brigham and Women’s Hospital in Boston.
More recently, Greenberg says, another type of dendritic cell was identified. These dendritic cells, called “plasmacytoid” (because they look like mature B cells called plasma cells), began to get attention in the mid-1990s, when biologists found they could produce huge amounts of substances called type 1 interferons, which include the potent cytokines interferon-alpha and interferon-beta.
Unlike myeloid dendritic cells, which only can pick up and present antigens, plasmacytoid dendritic cells also can respond to perceived invaders “immediately and locally,” Greenberg says. After they’ve responded by secreting type 1 interferons, they’re then believed to travel to lymph nodes, where they behave like myeloid dendritic cells.
 Recently, Greenberg and his colleagues detected what he calls the “signature” (characteristic pattern) of type 1 interferon activation in the muscles of patients with dermatomyositis.
Recently, Greenberg and his colleagues detected what he calls the “signature” (characteristic pattern) of type 1 interferon activation in the muscles of patients with dermatomyositis.
When cells are flooded with type 1 interferons, there are multiple downstream effects, mainly an increase in production of infection-fighting proteins from genes that go into high gear. When Greenberg looked at tens of thousands of genes in DM-affected muscle fibers, he found “90 percent of the highest expressed genes were genes that were induced [switched on] by type 1 interferons,” he says. When genes are switched on, cells read their DNA like a recipe and produce proteins from them. The DM-affected fibers were switching on genes and producing proteins as if they were fighting an infection, although no infection was apparent.
It’s been known for some 30 years that, in DM, muscle fibers themselves and the tiny blood vessels (capillaries) inside them are both injured. From this information, a hypothesis formed some 25 years ago, Greenberg says, that the damage to the capillaries and therefore blood supply came first and that damage to muscle tissue itself was a result.
“It’s a reasonable hypothesis,” he says, “but no evidence has emerged that this is actually the case.”
Greenberg has a different hypothesis, namely that the capillaries and muscle fibers are directly and similarly injured by interferon-induced proteins that they produce themselves.
“When you look at the tissues and stain them and ask where the proteins are, they’re present at the sites of the problem, in the capillaries and in the muscle fibers that are injured,” he says. “It looks like the capillaries and muscle fibers are producing interferon-induced proteins, and my hypothesis is that this may injure the muscle fibers and the capillaries when they do that.”
Greenberg thinks plasmacytoid dendritic cells are present in DM-affected muscles and says their presence has recently been confirmed in the juvenile form of the disease. They’ve also been found in DM-affected skin, he says.
Although scientific hypotheses and theories are important, Greenberg says, “it’s the patients that matter,” and the science is the means to help them.
“The most important thing is understanding the mechanism of a disease,” he says. “Understanding that there’s a substantial role for type 1 interferons in DM, and understanding what cell makes them, opens up a whole line of potential approaches.”
One approach would be to block interferon-alpha, and Greenberg says that’s being investigated by the biotechnology companies Genentech and Medimmune.
“One can go after [attempt to block] its production or secretion or processing,” he says, noting that he’s particularly interested in studying production of interferon-alpha at its source, the plasmacytoid dendritic cells.
“Understanding this further,” he says, “could lead to new treatment approaches.”
Supporting suppression
Although autoimmune diseases are widespread, most people go through life mounting immune responses to thousands of microbes and never develop an autoimmune problem.
In recent years, scientists have turned their attention to a less-explored part of the immune response: What keeps it from getting out of hand and ultimately turns it off?
MDA grantee Fu-Dong Shi at Barrow Neurological Institute in Phoenix is among them. Shi, a principal investigator in the Division of Neurology at Barrow with a special interest in myasthenia gravis, is looking at ways to expand the population of so-called regulatory T cells.
Shi says he hopes that expanding regulatory T cells will correspondingly suppress effector T cells (which spread an immune response), turning them down, “so they don’t produce cytokines and B cells won’t get help from them.”
His preliminary data from mice with MG shows that when the regulatory T-cell population reaches two to four times its usual level in these mice, their weakness improves. “It’s very exciting to see this happening,” says Shi, who hopes to try this strategy in patients with MG.
 Shi and colleagues are targeting a type of regulatory T cell known as the CD4+CD25+ cell because it bears surface markers CD4 and CD25.
Shi and colleagues are targeting a type of regulatory T cell known as the CD4+CD25+ cell because it bears surface markers CD4 and CD25.
The CD4+CD25+ cell is one subpopulation within the CD4+ cells, Shi says. Regular CD4+ cells are immune effector cells, which propagate an immune response and can cause autoimmune disease, he notes.
“But somehow within this family, there’s a small population that’s different, that has a suppressive function. We don’t know what the determining factor is, why these cells go in different directions when they mature.”
Some immunologists suggest that the relapses and remissions that occur in autoimmune disease may reflect the changing balance between effector cells and regulatory cells.
It seems that when immune responses are launched, a cytokine called interleukin 2 is produced by several types of cells, including T cells. Interleukin 2 can activate neighboring T cells that have up until then been dormant. But built into this system is a checkpoint: After the T cells have been active for a while, interleukin 2 normally kills them and expands the regulatory T cells, thus dialing down the immune response.
Shi and his colleagues at Barrow have been injecting MG-affected mice with interleukin 2 coupled with a compound that prolongs its survival in the bloodstream.
This combination significantly reduces the dosage of interleukin 2 required to increase the number of regulatory T cells, and it minimizes the potential side effects (such as fever and injection-site reactions) of using interleukin 2 alone, Shi explains.
He calls interleukin 2 a “drug in development” for suppressing immune responses. Other drug candidates also exist, he says, but they seem to have weaker effects.
“The possibility of suppressing autoimmunity would be very appealing to many patients,” Shi says. “It’s possible, but there’s a lot of hard work to be done in this area.”
The ultimate goal is to learn how to safely and effectively increase the number of regulatory T cells and maintain their survival long enough to suppress autoimmunity, all the while leaving intact the ability to fight infections.
“Before we can say we can use a cell-based therapy, we have to answer those questions,” Shi notes. “It will take time and effort, but for me, it’s a very appealing and natural therapy.”
Calming irritable cells from mom
Ann Reed, a former MDA grantee who studies and treats juvenile dermatomyositis at the Mayo Clinic in Rochester, Minn., agrees with Steven Greenberg’s hypothesis that dendritic cells are major trouble makers in this autoimmune disease (see “Interfering With Interferons” section above).
She thinks what makes these cells so irritable may have to do with where they come from — the patient’s mother.
During pregnancy, the placenta normally keeps maternal and fetal circulations separate. Small molecules can cross the placental barrier, but until recently, physiologists believed most cells from the mother stayed on one side of the placental barrier, and cells from the fetus on the other. Like many concepts in biology, this one recently has been overturned.
Starting in the late 1990s, researchers began observing that maternal and fetal cells jumped this supposedly impermeable wall much more often than previously presumed and that the phenomenon might be connected to the later development of autoimmune disease in either the mother or the child. (Fetal cells can end up in the mother, and maternal cells can end up in the child.)
 “My basic thought is that, if you look hard enough, you’ll find chimeric [originating in another organism] cells in everybody,” Reed says. “But you can see them more easily in autoimmune disease, because there are more of them.”
“My basic thought is that, if you look hard enough, you’ll find chimeric [originating in another organism] cells in everybody,” Reed says. “But you can see them more easily in autoimmune disease, because there are more of them.”
Reed and her colleagues published a study in 2004 that found maternal cells, mostly the dendritic type, in 60 out of 72 (83 percent) of children with juvenile DM; 11 out of 48 (23 percent) of their unaffected siblings; and five of 29 (17 percent) of unaffected, unrelated children.
She believes “something is disrupted,” probably in the placenta, that allows more cells than normal to transfer from mother to fetus. The development of autoimmunity, Reed says, is probably related to the dose of foreign cells.
But it also seems to require what Reed terms a reactive environment, an “environment that’s ripe for a cell that might be hanging out to get stimulated” even if it’s been around for a long time without causing a problem.
“Something has to happen in the local environment in the tissue to make this start, something like inflammation or infection,” Reed says. “Most of the time, these settle down. But there’s something there that allows for that sort of irritation to keep going and get amplified in these patients. We think dendritic cells play a key role. They’re out there on patrol, and then they’re maturing and calling in everyone else to help them. An environment starts setting itself up as if there’s a problem.”
In both adults and children with DM, Reed says, “we see enormous numbers of plasmacytoid dendritic cells. They stimulate interferon, which stimulates other things downstream.”
Reed says it might be interesting to think about a targeted antibody that removes certain kinds of cells or blocks those cells’ ability to respond. Creating an antibody to a maternal immune-response protein or blocking receptors on maternal dendritic cells are two thoughts she’s had. She cautions, however, that once the immune response gets started, it might be hard to stop it with this kind of strategy.
Better and earlier diagnosis would help, and Reed also is working on that, by analyzing molecular signatures, such as whether protein production from certain genes is turned up (upregulated) or turned down (downregulated), in various diseases. She’s found that DM, polymyositis and inclusion-body myositis all have different signatures.
“We’re looking at key patterns of gene upregulation or downregulation to see whether they’re involved in causing inflammation,” she says. “We think there are patterns that are causing the body to turn down a certain pathway. A cell with a certain stimulus will go one way or another way.” Ideally, it could be steered in the right direction.
Removing antibodies
In the late 1970s, a young physician named Peter Dau at Children’s Hospital of San Francisco received MDA support to develop what many at the time thought was a foolish strategy to treat MG: mechanically removing disease-causing antibodies. (In London, John Newsom-Davis, David Keith Peters and others also were pursuing this approach.)
But Dau persisted and applied “plasmapheresis” to sift out antibodies to acetylcholine receptors that were presumed to be causing the disease.
On a regular basis and in conjunction with immunosuppressant medication, patients had their blood run through a filter that separated the liquid component (plasma) from the blood cells. The autoimmune antibodies, along with needed antibodies and other small but necessary proteins, such as clotting factors, were thrown out with the plasma, and the cells were returned to the patient, along with replacement plasma. The treatment, though somewhat risky, often led to sustained remission of the disease.
The average MG patient needed about nine weekly plasmapheresis treatments, although Dau had one patient who required 103.
Later modifications reduced the removal of or replaced plasma proteins other than antibodies, but even so, antibodies needed for infection fighting are depleted, at least temporarily.
The strategy is still used in MG today, allowing some patients to reduce their intake of toxic immunosuppressant drugs and providing rapid treatment in an emergency.
MDA grantee Socrates Tzartos at the Hellenic Pasteur Institute in Athens, Greece, would like to see plasmapheresis made more useful by refining it to sift out only the autoimmune antibodies.
Fortunately, the antigens (specific parts of the acetylcholine receptor) and the antibodies are better understood in MG than they are in many autoimmune diseases, and Tzartos has been able to apply that knowledge to his strategy. (In fact, Tzartos contributed considerably to mapping the acetylcholine receptor when he was a postdoctoral student some 30 years ago.)
In 1994, Tzartos’ group began making fragments of acetylcholine receptor proteins to use as molecular “adsorbents” (molecular magnets) that would attract and stick to acetylcholine receptor antibodies. It took until 2001 to see the first successful immunoadsorption experiments.
He’s now successfully created adsorbents that filter out several types of antibodies taken from patients with MG; namely antibodies to the alpha, beta, gamma and epsilon subunits (parts) of the acetylcholine receptor.
In contrast to standard plasmapheresis, Tzartos says, his group’s approach uses columns with attached fragments of the acetylcholine receptor that bind and eliminate from the plasma only the disease-related, autoimmune antibodies.
 “We hope in a future, improved version of our approach, all autoantibodies may be depleted from the patient in a single session,” Tzartos says. “Of course, later production [by the immune system] of new autoantibodies cannot be avoided.”
“We hope in a future, improved version of our approach, all autoantibodies may be depleted from the patient in a single session,” Tzartos says. “Of course, later production [by the immune system] of new autoantibodies cannot be avoided.”
He estimates it will take a year or two to move his device from the laboratory to the clinic. “The good thing is that it’s not a drug,” he notes. “Nothing new is to be administered to the patient, so it should more easily go to the clinic.”
Tzartos believes the setup he has now will target about 20 percent to 30 percent of MG patients with moderate to severe symptoms. But, he says, “we are also working hard toward broadening its appropriateness to cover most myasthenic patients, and to make the procedure simpler.” He adds that it also may have application to other autoimmune diseases in which antibodies play a major role.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
