
Through the Looking Glass with FSH Dystrophy Researchers
Impossible things
In 1990, Sara Winokur was a doctoral student in the laboratory of John Wasmuth, a professor of biological chemistry and a prominent researcher in genetics at the University of California at Irvine.
It was an exciting time in genetics. The genes that, when mutated (flawed), cause diseases, were rapidly being identified. Among the first, in 1986, had been one for Duchenne muscular dystrophy.
By late 1990, researchers knew that something on chromosome 4 was going wrong in people with facioscapulohumeral dystrophy (FSHD), a type of MD that showed a preference for the facial, shoulder and upper arm muscles.
Winokur recalls Wasmuth’s enthusiasm for finding the FSHD gene. (Wasmuth died unexpectedly in 1995 at the age of 49.)
"This is a great project for you," he told her, "because now it’s been mapped to chromosome 4. In another three or four years, you’ll know what the mutation is, what the gene is and what the problem is."
"I thought it sounded great," Winokur says. "So I started to work on the project.
"Here it is [more than] 15 years later, and, although we’ve come a long way, the specific genes involved have not been clearly identified."
The chromatin connection
Although chromosomes are usually thought of as strings of DNA, they’re actually DNA intertwined with protein molecules. Together, these are known as chromatin, a term coined in the 19th century that simply means "colored material" and describes how chromosomes looked under the microscopes of the day.
 In areas where chromatin is tightly compacted and condensed, genes are inactive. Where it’s expanded and open, genes are active, meaning the cell’s machinery is using them, by following DNA instructions, to make proteins. (The scientific term for the open type is euchromatin; for the closed type, it’s heterochromatin.)
In areas where chromatin is tightly compacted and condensed, genes are inactive. Where it’s expanded and open, genes are active, meaning the cell’s machinery is using them, by following DNA instructions, to make proteins. (The scientific term for the open type is euchromatin; for the closed type, it’s heterochromatin.)
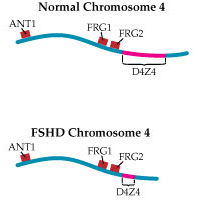
By the mid-1990s, understanding of the chromosome 4 region associated with FSHD — later dubbed D4Z4 — had progressed considerably.
Investigators had found that, in that region, there are a number of repeated sections of DNA, each about 3,200 molecules long, placed end to end. Normally, the D4Z4 region contains 11 to 150 of these repeats, while FSHD-associated chromosomes have fewer than 11. Later work would confirm early suspicions that, despite the clear linkage of the disease to this area, there are no actual genes inside D4Z4.
It was time to start imagining, if not yet quite believing, impossible things.
Wasmuth and others proposed that loss of a piece of chromosome 4 might shift nearby genes from a region of open chromatin into a region of condensed chromatin, thereby silencing normally active (expressed) genes. This type of mechanism is called a position effect.
Researchers continue to explore chromatin changes as a possible mechanism in FSHD. Recently, MDA grantee Kyoko Yokomori, at the University of California at Irvine, found that the D4Z4 region normally contains both the open and the condensed forms of chromatin.
"It was a little bit different from what people predicted," Yokomori says. "It’s actually a mixture. We don’t know how it’s actually organized.
"The important thing is that the chromosome 4 D4Z4 repeat has both the heterochromatin and euchromatin structures. When we looked at FSHD patients’ cells, we found that in all cases this heterochromatin structure seems to be specifically lost, while the euchromatin structure is OK."
This observation suggests that abnormal gene expression, if there is any, is in the direction of activating, rather than silencing, genes, as Wasmuth had proposed.
Yokomori says she thinks chromatin abnormalities and gene activation have something to do with FSHD, but she doesn’t think the problem is confined to local effects on chromosome 4.
"Whatever the mechanism is, probably it’s not a simple spreading of the heterochromatin to the neighboring regions," she says. "It could affect completely different chromosomes. This is still a model, so it may turn out to be wrong, but there are several reports now saying that a regulatory region on one chromosome can interact transiently with genes located on a completely different chromosome."
Pushing the envelope
Sara Winokur completed her doctoral studies in 1995 and has been an MDA research grantee since 1999. She believes that alterations in chromatin structure may affect genes near D4Z4. But she also believes something else is going on.
"We’re proposing that changes in local chromatin structure, which do exist, could affect interactions with the nuclear envelope," says Winokur, referring to two membranes that surround each cell nucleus.
For many years, scientists believed that the role of the nuclear envelope was to restrict passage of various molecules into and out of the nucleus, where the chromosomes reside. But recent findings suggest there may be more to the envelope than this traffic cop function.
"There’s a class of muscular dystrophies that involve nuclear envelope proteins," Winokur says, referring to Emery-Dreifuss MD and one form of limb-girdle MD. "How do they come about? We don’t really know the answer to that, but one hypothesis is that those nuclear envelope proteins somehow regulate gene expression.
"We’re suggesting that, since this chromatin region clearly localizes to [is located at] the nuclear envelope, it’s certainly possible that there are alterations in how that chromatin can interact with the nuclear envelope. The chromatin itself has changed, and therefore the interactions with the nuclear envelope might be different.
"Local genes may well be affected," she says, "but it could be that, because this region is now in a particular compartment in the nucleus, and there are other regions of the genome that also localize to this compartment, there may be interactions between the FSHD region and those other regions. So that’s how it may be that there’s a broader effect."
So far, Winokur’s group has found that a section of chromosome 4 near D4Z4 normally anchors itself to the nuclear envelope.
She hastens to add, "We have not detected a difference in localization between the FSHD and the normal cells. But we’re still proposing that because of the altered chromatin structure, it’s still not interacting properly."
Along with Yi-Wen Chen and MDA grantee Eric Hoffman, both at Children’s National Medical Center in Washington, Winokur has compared gene activity in normal versus FSHD-affected muscle, using a technique called expression profiling.
A pattern seems to be emerging. "When we did the expression profiling, we found a number of genes that are involved in myogenic differentiation [maturation of nonspecific cells into muscle cells] that seem to be different in FSHD versus normal muscle."
In addition, the FSHD-affected cells seem to have the same pattern of gene activation as do cells affected by the two known nuclear envelope forms of dystrophy.
"It broadens the pool of genes that we should be looking at," Winokur says. "[FSHD] is probably going to turn out to be an overexpression or inappropriate expression of genes that should be silenced in the mature, differentiated muscle fiber."
Diving into D4Z4
Rossella Tupler, at the University of Massachusetts-Worcester and Universita degli Studi di Modena e Reggio Emilia in Modena, Italy, has a different view, although she too believes aberrant gene regulation underlies FSHD. (Tupler was an MDA grantee from 1999 through 2004.)
In 1992, she was working in a genetics lab and had collected samples from people with relatively large chromosomal abnormalities involving the tip of chromosome 4. Reasoning that comparing the missing chunks of chromosome 4 with the patients’ symptoms might lead her to the FSHD gene, she eagerly got to work. It proved a dead end.
"We analyzed the patients, and the only thing I found was that the deletion of the region did not cause any [signs of] FSHD. So from my observation and my experiments, the disease was not associated with a lack of something.
"In that period," she says, "I received a small grant, and I started collecting FSHD patients and families. Back then we were of course looking for genes, but at a certain point, I thought, ‘This can really be something related to abnormalities in gene expression.’"
Tupler compared muscle samples from people with and without FSHD and noted that expression of the ANT1 gene was higher in FSHD-affected than unaffected muscle tissue.
"That was before the human genome sequencing, so there was much less information," she says. "It was a completely different time."
Failing to find genes in the D4Z4 region, where the FSHD mutation was thought to be, she reconsidered the facts. "I thought, I have to run an experiment in which I can understand if D4Z4 by itself has a function.
"It was March 1999. I was in the lab, and I asked this colleague, ‘Which kind of experiment can I run to see if D4Z4 has a role in [activation]?’ And this colleague said, ‘Try a band shift [a lab technique].’ And I did."
What Tupler saw with the band shift were proteins sticking to D4Z4. "From there," she says, "I started a completely new story."
Variations in chromatin may, Tupler believes, account for the variable severity of FSHD and for its selection of particular muscle groups. But she doesn’t think chromatin structure is the main issue.
All about FRG1?
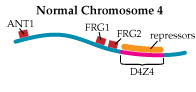
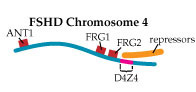 In Tupler’s view of FSHD, the loss of DNA repeats in D4Z4 makes the region too small to play its role as a landing pad for a “repressor complex,” a group of proteins that she says normally attaches to D4Z4 and shuts down nearby gene expression. In 2002, she and her colleagues published findings that strongly support this hypothesis.
In Tupler’s view of FSHD, the loss of DNA repeats in D4Z4 makes the region too small to play its role as a landing pad for a “repressor complex,” a group of proteins that she says normally attaches to D4Z4 and shuts down nearby gene expression. In 2002, she and her colleagues published findings that strongly support this hypothesis.
Then, last year, her group published a paper in the prestigious journal Nature that implicates overactivation of a gene called FRG1 as a crucial factor in, if not the only cause of, FSHD.
The researchers bred mice that overproduced proteins from three genes lying near the D4Z4 region on chromosome 4. Those that overproduced the FRG1 protein (with no other defect) developed a disease that Tupler says closely resembles FSHD; and the more FRG1 they produced, the worse the disease was.
“We generated mice overexpressing ANT1, FRG1 and FRG2,” Tupler says. “And clearly, overexpression of FRG1 causes muscular dystrophy. What is very interesting is that those mice have several features that are similar to the human disease.”
She summarizes her findings this way. “This is my vision of the disease: We have a repetitive element. The repetitive element binds to something that is repressing expression. But then when we remove the DNA element that binds this complex, we have the overexpression of genes.
“Among those genes there is one gene, FRG1, which controls a number of other genes. And this is a kind of cascade effect.”
There’s only one problem with Tupler’s hypothesis: Other researchers of equal reputation have found no increase in FRG1 expression. Tupler says the discrepancy is related to differences in experimental methods. It remains, however, unresolved.
Which way from here?
Tupler’s group is already working on identifying specific treatments for people with FSHD.
“We’re screening for small molecules that can suppress the activity of FRG1, so it’s a pharmacological approach,” she says. “We’re using molecules that are working on the protein or on the cell. We started the screening last year, and it apparently is working well.”
Winokur believes more of the basic biology remains to be figured out before treatments can be contemplated.
“The problem is that we still don’t know clearly which genes [are involved],” she says. “Until we know which specific genes are affected by any of these mechanisms, we cannot develop effective therapies.
“It’s most likely going to be the case that in FSH dystrophy we actually need to utilize therapies to silence a gene. Currently there isn’t anyone working on that for FSHD. There are a number of groups that are becoming interested in doing so, but we’ve always been limited by not knowing precisely which genes to look at.”
From Funny Smile to Forced Retirement
Living with FSH dystrophy
Like many people with FSH dystrophy, Robert Mingo would wait until well into adulthood before getting a diagnosis of his condition, although in retrospect, the clues were there all along.
First, there was the facial weakness that made photo sessions an ordeal.
“I’ve got a funny smile,” says Mingo, now 45 and living in Minnetonka, Minn., with his wife, Amy. “I’d go into school photos, and my mom would say, ‘You better not have that stupid grin on your face. Show your teeth.’ Sometimes I could, but most of the time that stupid grin was on my face.”
Shoulder weakness also came on early. “I learned how to throw a Frisbee differently,” Mingo says. “Just about anything I learned how to do, I had to do different.”
Gradually, Mingo’s weakness progressed. “My upper body’s weaker than my lower body,” he says. “I can’t get my elbows higher than my shoulders. I struggle with washing my hair and combing my hair and brushing my teeth. I don’t even read books, because I don’t like to hold the book up.”
For 10 years, he worked as a veterinary assistant, until increasing weakness caught up with him.
“I kept slipping on the wet floor. I couldn’t lift the dogs anymore. I just didn’t have the strength for the job,” Mingo says. “Part of me thought I was just lazy and didn’t work out hard enough. I didn’t want to tell anybody I was getting weaker, because I thought it was my fault.”
In spring 2002, Mingo was 40, recently married, working in a conven-ience store and distraught about the insidious progression of his weakness.
 “I was trying to fill coolers. I was trying to do everything. I was miserable. I was in so much horrible pain, but I didn’t want to tell anybody, because everybody’s got pains. I just figured I’d be a whiner if I complained,” he says. “When I told my job, they told me they couldn’t use me anymore.”
“I was trying to fill coolers. I was trying to do everything. I was miserable. I was in so much horrible pain, but I didn’t want to tell anybody, because everybody’s got pains. I just figured I’d be a whiner if I complained,” he says. “When I told my job, they told me they couldn’t use me anymore.”
A visit to his family doctor, followed by a referral to a local neurologist and then to the MDA clinic at the University of Minnesota Medical Center in Minneapolis, gave him a diagnosis at last: facioscapulohumeral muscular dystrophy.
Although a clear reason for his weakness gave him relief from blaming himself, it also heralded the start of what Mingo calls his “big, black cloud” year. “It was a desperate year,” he recalls. “I couldn’t work. We filed for bankruptcy.”
By 2003 Mingo was using a wheelchair much of the time.
“We used a push chair for a while, and then MDA came and rescued me,” he says. “They brought me a [power] chair and loaned it to me until we could get insurance and doctors and everybody working together. My pride was a big issue, but once I learned just how much better it was, how I wasn’t tired all the time, it wasn’t such a bad thing.”
Although respiratory muscle weakness requiring breathing support is extremely rare in FSHD (a Dutch study estimated it at 1 percent of patients), doctors diagnosed it in Mingo and prescribed air under pressure (BiPAP) by mask during sleep.
These days, Mingo sells rare coins on the Internet, while Amy works at a bank.
They’ve had to give up some of their goals, he says. But, “Life is good. It really is. Sometimes you gotta find a new normal and a new way to approach things.”
His support group is “the MDA staff and all the events we go to. We go to the balls and the dances. I used to love to dance when I could get my legs underneath me and actually dance. Now if I tried to dance outside the wheelchair, I’d probably wind up on my face real quick,” he says.
“So now I’ve learned how to dance in a wheelchair, and it’s just as fun — well, not just as fun, but it’s close.”
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
