
Progress Now Fall 2015

Research and clinical trial updates
Towards Treatments and Cures
New grants pave the way to progress
As part of an effort to refocus its research grants program, MDA began funding 36 new research and development grants, totaling nearly $10 million, on Aug. 1, 2015. The awards were made to researchers seeking to increase understanding of and find promising treatments and cures for muscular dystrophy, ALS and related neuromuscular diseases that limit strength and mobility. Making up the summer 2015 grant cycle, the awards reflect an increase in spending within MDA’s research program and underscore MDA’s commitment to investing in research across diseases — with the expectation that increased understanding in one disease area will inform progress and lead to research and medical breakthroughs in other disease spaces.
MDA’s Medical and Scientific Advisory Committees, comprising some 40 of the world’s leading clinicians and scientists who serve in these volunteer roles for MDA, recommended the top grants out of 350 applications to MDA’s Board of Directors. Board approvals were made in July.
Among the research projects funded by the new grants:
- Scientists at the University of Kentucky in Lexington will study a new drug target for ALS and test a potential therapy.
- Scientists at Biosciences Institute, University of São Paulo (Brazil), will study how a protein called Jagged1 prevents the symptoms of Duchenne muscular dystrophy (DMD) in animal models bearing the mutation that causes the disease.
- Scientists at the University of Oklahoma Health Sciences Center will work to find the optimal HDAC inhibitor (and dose) for increasing production of the frataxin protein, which is deficient in Friedreich’s ataxia (FA) and, in parallel, test an alternative HDAC inhibitor that may prove more effective than others tested to date.
- Scientists at Columbia University in New York will study the underlying mechanisms that drive a recently discovered form of congenital muscular dystrophy (CMD) and test three possible treatment paradigms.
- Scientists at Johns Hopkins University School of Medicine in Baltimore will test a gene therapy strategy to increase SMN protein levels in
spinal muscular atrophy (SMA). This project is co-funded by the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM), as MDA and AANEM continue to build on a partnership aimed at providing neuromuscular disease education and supporting research related to muscle and nerve disorders.
The new grants complement a wide range of ongoing neuromuscular disease research MDA is supporting worldwide.
ALS (amyotrophic lateral sclerosis)
Q on the Move
FDA clears way for clinical trial to test ALS cellular therapy
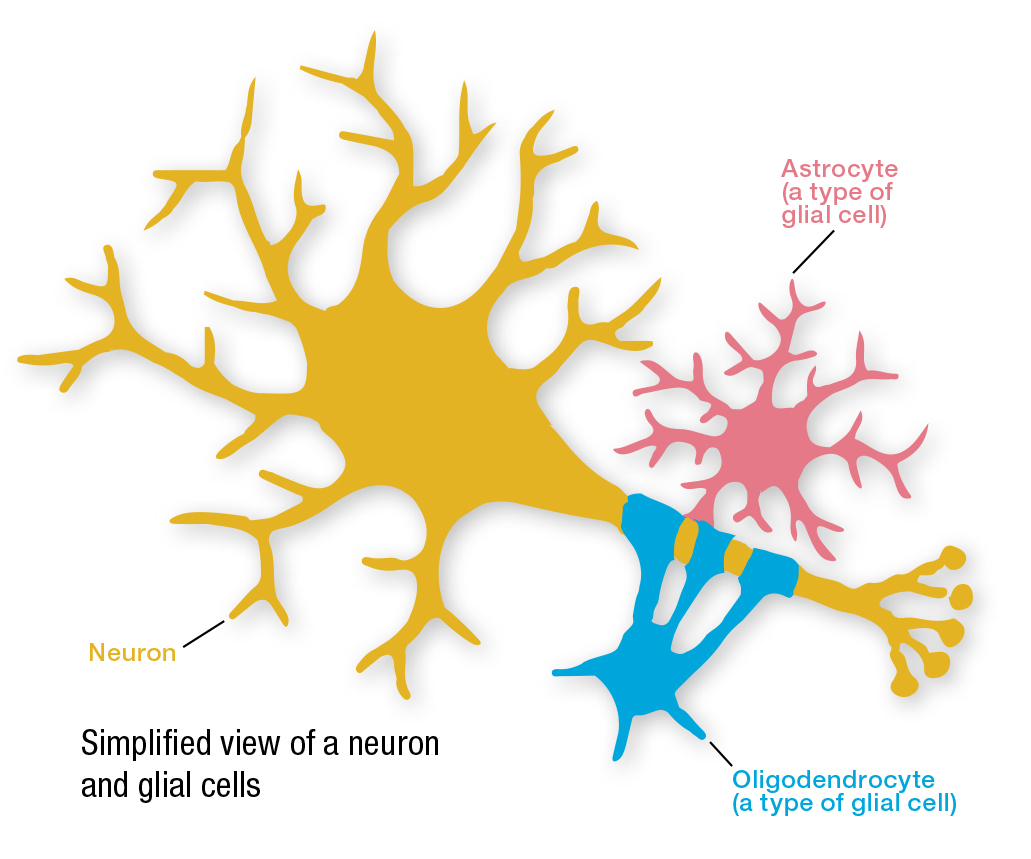
Q Therapeutics announced in June that the U.S. Food and Drug Administration (FDA) has cleared its Investigational New Drug Application (IND) for the initiation of phase 1-2a clinical trials of its experimental cellular therapy, Q-Cells, in people with ALS (amyotrophic lateral sclerosis).
Q-Cells are human glial-restricted progenitor cells (GRPs) that give rise to two types of specialized glial cells — astrocytes and oligodendrocytes — which are critical for normal motor neuron function.
The trial, which is set to begin in October, will test the safety, tolerability and early efficacy of Q-Cells transplantation in 12 participants with ALS. The trial is not yet open for enrollment.
The FDA has previously granted Q-Cells orphan drug status — a designation intended to advance the evaluation and development of therapeutics that demonstrate promise for the treatment of rare diseases — for ALS.
To find out more about this trial, search for “NCT02478450” at ClinicalTrials.gov.
Duchenne muscular dystrophy (DMD)
Moving Forward
Corticosteroid treatment deflazacort advances toward approval in the U.S.
Marathon Pharmaceuticals has begun the New Drug Application (NDA) process for deflazacort as a treatment for Duchenne muscular dystrophy (DMD).
Deflazacort, a corticosteroid, works as an anti-inflammatory and immunosuppressant. It has been shown to slow the loss of muscle strength and function, preserve cardiac and respiratory function, and reduce the incidence of scoliosis (curvature of the spine) in people with DMD. Importantly, the unwanted side effects often experienced with corticosteroids, such as weight gain, loss of bone mass, glucose intolerance (diabetes) and behavioral issues, may be less severe with deflazacort as compared to other steroids.
Marathon expects to submit the NDA to the FDA in the first quarter of 2016. If the application is approved, the company could make the drug commercially available in the United States in the first quarter of 2017.
To learn more about deflazacort in DMD, read Charting a Corticosteroid Course: The Benefits and Side Effects of Prednisone and Deflazacort.
Trial Seeks Participants
CAT-1004 to be tested in boys with DMD, ages 4 to 7
A phase 1-2 clinical trial called MoveDMD is open for recruitment at Shriners Hospitals for Children in Portland, Ore., and the University of Florida in Gainesville, Fla., with a third site expected to open soon in Philadelphia.
The two-part trial will test the experimental therapy CAT-1004 in boys with DMD ages 4 to 7 years old. Investigators are looking for a total of 18 boys to participate in the first part of the trial, which will examine how the drug moves through the body and collect data on physical function and muscle strength of the lower and upper legs to help understand potential benefits of the drug in DMD.
CAT-1004, in development by Catabasis Pharmaceuticals, is an anti-inflammatory compound that is designed to act similarly to corticosteroid medications (like prednisone and prednisolone) but with fewer unwanted side effects. Preclinical data suggest that CAT-1004 may reduce muscle damage, stress and inflammation.
Boys who participate in the first part of the trial and who meet study criteria will be asked to participate in the second part of the trial, which will further evaluate safety and efficacy of the experimental therapy.
To learn more about this trial, including inclusion and exclusion criteria, search for “NCT02439216” at ClinicalTrials.gov.
Treatments on the Horizon
Two exon-skipping drugs to treat DMD in a subset of boys diagnosed is poised to enter U.S. market
BioMarin announced June 29 that the FDA has accepted its New Drug Application (NDA) for drisapersen, and Sarepta announced Aug. 25 that the FDA has accepted its NDA submission for eteplirsen. Both “exon-skipping” drugs target exon 51, and may help up to 13 percent of DMD patients. Exon skipping is a treatment strategy in which sections of genetic code are “skipped,” allowing the creation of partially functional dystrophin, the muscle protein missing in DMD. Exon skipping is not a cure for DMD, but it could lessen the severity of muscle weakness and atrophy that are the hallmark of this disease. Both companies hope to have an FDA decision by early 2016.
Strong Prospects
Early results for HT-100 in DMD are encouraging
The experimental DMD drug HT-100, in development by Massachusetts-based Akashi Therapeutics with support from MDA, has shown encouraging preliminary results in an ongoing phase 1b-2a clinical study in boys with DMD. Data from this ongoing clinical trial shows that boys with DMD who were treated with HT-100 demonstrated improvements in muscle strength. In addition, the drug continues to be safe and well-tolerated by trial participants.
MDA has contributed more than $1 million toward the development of HT-100 as a therapy for DMD, supporting early-stage work that informed the laboratory development of the drug and providing funding for the ongoing trial as well.
HT-100 is an oral compound that is intended to reduce inflammation and scar tissue formation and promote regeneration in DMD-affected muscles. It does not target specific mutations in the dystrophin gene, making it potentially useful for all patients with DMD.
For more on this trial, read DMD: Multicenter Trial to Test Drug that Fights Muscle Scarring, Inflammation.
Fighting Fibrosis
FG-3019 to be tested in DMD
FibroGen announced in July that it has received FDA clearance to begin clinical testing of FG-3019 in DMD. The company plans to initiate a multisite phase 2 trial in late 2015 to test the therapy in nonambulatory patients.
In DMD muscle cells, an absence of the dystrophin protein required for normal muscle fiber function leads to muscle damage, inflammation, fibrosis (scarring), and progressive dysfunction and weakness. In preclinical studies, FG-3019 reduced muscle fibrosis and significantly improved muscle function. Separately, in clinical trials for another fibrotic condition, idiopathic pulmonary fibrosis, FG-3019 was shown to reverse fibrosis in a significant proportion of patients.
If testing is successful, this therapy could help improve the lives of people with DMD by delaying loss of muscle function.
Facioscapulohumeral muscular dystrophy (FSHD)
Quality of Care
Experts publish first-ever evidence-based guideline for FSHD diagnosis and care
The American Academy of Neurology (AAN) and American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) have released a guideline for the diagnosis and care of people with facioscapulohumeral muscular dystrophy (FSHD), a genetic muscle disorder in which muscles of the face, shoulder blades and upper arms are among the most affected.
Based on the best available scientific studies of FSHD, this is the first-ever evidence-based guideline to help physicians best manage the care of people with FSHD. It includes key recommendations on:
- genetic testing to confirm diagnosis; and
- management of FSHD-associated complications, including those related to lung and breathing function, shoulder problems, hearing loss and pain.
Goals for the new guideline are to increase awareness that although cures are not currently available, disease management is important and valuable; raise awareness of the strong need for further FSHD research; and increase awareness of the importance of genetic testing for accurate diagnosis.
Read the guideline in the Guidelines section on aan.com, or view a family-friendly version. To learn about pain management, read Medication, Meditation and Movement.
Hereditary inclusion-body myopathy (HIBM)
Advancing Ace-ER
Extended release aceneuramic acid in HIBM is being tested in a phase 3 trial
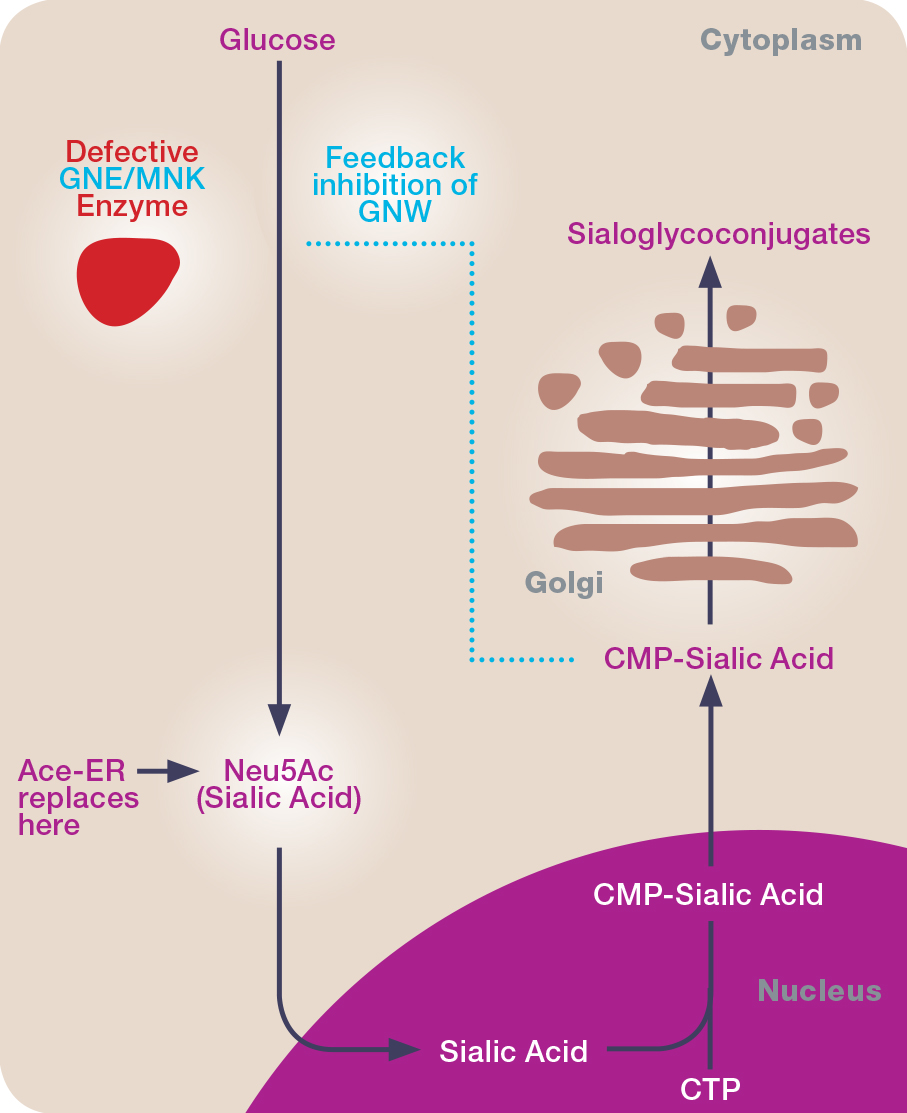
Ultragenyx Pharmaceutical has initiated a phase 3 study of aceneuramic acid extended release (Ace-ER) tablets for the treatment of GNE myopathy, also known as hereditary inclusion-body myopathy (HIBM) and Nonaka distal myopathy.
The new placebo-controlled clinical study, conducted in approximately 80 participants, will assess the efficacy and safety of six grams per day of Ace-ER over 48 weeks. The primary endpoint of the study is a composite of upper extremity muscle strength as measured by handheld dynamometry (HHD). Key secondary endpoints include the GNE Myopathy-functional activity scale (GNEM-FAS), a disease-specific, patient-reported outcome that measures mobility, upper-extremity function and other indicators of lower-extremity muscle strength.
To learn more about this trial, including inclusion and exclusion criteria, search for “NCT02377921” at ClinicalTrials.gov.
Limb-girdle muscular dystrophy (LGMD)
Diagnostic Accuracy
Additional funding will expand access to genetic testing for LGMD
Genzyme has provided $700,000 in additional funding to continue a limb-girdle muscular dystrophy (LGMD) genetic testing program launched in March. The program, which allows MDA clinics to offer genetic testing to people experiencing muscle weakness suspected to be caused by LGMD and who do not already have a genetic diagnosis, has received an overwhelming response. This additional funding will allow more people living with LGMD, and their clinicians, to find the most accurate diagnosis and treatment paths available.
Since the partnership began in March, more than 600 tests have been administered in MDA clinics across the country.
LGMD is a disorder with more than 30 genetic subtypes, all of which cause weakness in the limb-girdle muscles — the muscles surrounding the shoulders and hips — and some of which are much more likely than others to involve the cardiac or respiratory muscles. This test will allow clinicians to provide accurate diagnosis and treatment.
The test requires a saliva sample, which can be collected at any one of the nearly 200 MDA clinics across the country and is then sent to Emory Genetics Laboratory in Decatur, Ga. Results are returned to the clinic physician and communicated to patients and their families within two to three months.
Periodic paralysis
FDA approves first medication for hyperkalemic and hypokalemic periodic paralysis
Taro Pharmaceuticals announced in August that the FDA has approved Keveyis for the treatment of hyperkalemic and hypokalemic periodic paralysis. Keveyis is the first medication to be approved for the treatment of this group of hereditary disorders that causes episodes of muscle weakness or paralysis. Keveyis is expected to be available for patients in late 2015, and Taro is working to make sure people diagnosed with periodic paralysis can receive treatment as soon as possible.
MDA provided support for the development of Keveyis beginning in the early 1990s, including contributions toward the early-stage development of the drug and funding support for a phase 3 human clinical trial — all of which has helped make this new treatment possible.
To be notified when the drug becomes available, enroll at keveyis.com.
Spinal muscular atrophy (SMA)
Positive Progress
Gene transfer therapy chariSMA demonstrates encouraging results
Biotechnology company AveXis announced promising interim results in June for its ongoing phase 1 SMA gene therapy trial in infants with type 1 spinal muscular atrophy (SMA).
AveXis reported that all three infants being treated with a low dose of the company’s SMA drug, known as chariSMA, have surpassed the age at which untreated babies with type 1 SMA lose their battle with the disease. The three currently range in age from 15 to 18 months. All nine infants enrolled in the trial remain alive, and none have progressed to the need for permanent ventilation (defined as 16 hours/day or greater use of ventilation assistance).
SMA is caused by a mutation in the SMN1 gene, which results in a deficiency of SMN protein.
ChariSMA contains the gene for the full-length SMN protein, encased in the shell of a type 9 adeno-associated virus (AAV9 “vector”) that serves as a “gene transfer” or “gene therapy” delivery vehicle.
The phase 1 trial of intravenous SMA gene therapy is being conducted at Nationwide Children’s Hospital in Columbus, Ohio, under the direction of neurologist Jerry Mendell, a longtime MDA research grantee and co-director of the MDA clinic at his institution. (MDA is not, however, funding this trial.)
For more information on chariSMA, search for “NCT02122952” at ClinicalTrials.gov.
Embracing Opportunity
A multicenter phase 2 study of ISIS-SMNRx seeks participants
Biogen, in partnership with Isis Pharmaceuticals, has initiated a new phase 2 clinical trial, called EMBRACE. This multicenter phase 2 clinical study aims to test the safety and exploratory efficacy of the investigational drug ISIS-SMNRx in approximately 20 patients with infantile or childhood-onset SMA over a 14-month period.
ISIS-SMNRx is being developed by Isis Pharmaceuticals in collaboration with Biogen. Developed using a gene-modifying strategy known as antisense, it targets the SMN2 gene and encourages production of the full-length SMN protein that is needed but deficient in SMA-affected cells.
EMBRACE is the fourth ongoing trial testing the same drug (ISIS-SMNRx) for SMA patients. The other trials are ENDEAR, CHERISH and NURTURE. The purpose of the new trial is to capture patients who do not meet the age requirements or other criteria used in the other trials.
To learn more about how to participate in this trial, search for “NCT02462759” at ClinicalTrials.gov.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
