
In Focus: Myotonic Muscular Dystrophy

A challenging and multifaceted disease
As far back as Carla Licon can remember, her mother had difficulty opening jars and walking long distances. Licon’s mother wore ankle braces, and she also had an unusual symptom known as "myotonia," the inability to relax muscles, such as a clenched hand, at will.
Licon, who is 31 and lives in Victoria, Texas, thinks these symptoms started when her mother was in her 20s. Later, her mother’s respiratory muscles weakened, leading to pneumonia, respiratory failure and her death at the age of 52.
The diagnosis was myotonic dystrophy, also known as myotonic muscular dystrophy and dystrophia myotonica, and abbreviated as either MMD or DM.
Licon’s grandfather, a career military man, also had the disease. His MMD caused him to be put on light duty in his later years, and then to retire. He too died of pneumonia, before Licon was born.
When Licon was almost 17, she started noticing that her hands would start "locking up," and she began feeling tired. She explained it by saying she had worked too hard, but the symptoms gradually worsened. "It really didn’t dawn on me until later that it was actually the muscular dystrophy," she says.
About a decade later, she began noticing leg cramps and spasms in her limb and abdominal muscles.
Then things "seemed to go downhill pretty quick," Licon says. In early 2011, she had to leave her job at a phone company call center. “It got to where I couldn’t work anymore,” she says. "I couldn’t function during the day. My hands would lock up, and I wasn’t able to type in the notes that I needed to. On occasion my throat will kind of cramp up to where you can’t understand anything I’m saying. I have to stop talking for a little bit and wait for it to get back to normal, and that’s no good when you’re on the phone all day."
Recently, Licon has had to move out of her apartment and into her future mother-in-law’s home. "The apartment where we were living has stairs, and it was getting to where I was falling at least once a week," she says.
Adult-onset MMD1 affects multiple systems, organs
Licon has what’s now known as adult-onset type 1 MMD (MMD1), a genetic disease that’s caused by an expanded section of DNA on chromosome 19. The adult-onset form — sometimes called "classic" myotonic dystrophy and thought to be the most common type — has its onset in late adolescence or young adulthood. It affects a number of body systems, although there is a wide range of severity.
The muscle-related symptoms include myotonia, for which the disease is named, and weakness, particularly of the face, neck, and limb muscles that are furthest from the center of the body (the distal muscles), such as the forearms, hands, lower legs and feet. Over time, all limb muscles can become weak.
Among the most serious effects of MMD1 are weakness of the breathing and swallowing muscles and dysfunction of the heart muscle, particularly the tissue in the heart that conducts electrical impulses from one part of the heart to another.
 The so-called "smooth" muscles of the gastrointestinal tract can be affected, causing diarrhea, constipation and abdominal pain. Other smooth muscles that line the hollow organs of the body, such as the uterus and gallbladder, can be affected as well, leading to obstetric complications and gallstones.
The so-called "smooth" muscles of the gastrointestinal tract can be affected, causing diarrhea, constipation and abdominal pain. Other smooth muscles that line the hollow organs of the body, such as the uterus and gallbladder, can be affected as well, leading to obstetric complications and gallstones.
The lenses in the eyes almost always develop cataracts, which can be surgically removed when they interfere with vision. The cataracts are distinctive and have been described as resembling Christmas tree lights.
And then there are the effects on the brain, causing a range of symptoms, including learning disabilities, difficulty with decision-making, and what some psychologists have called an "avoidant" or "apathetic" personality type.
Excessive daytime sleepiness and chronic fatigue are among the most puzzling and troublesome of MMD1 symptoms, and their origin appears to be complex and probably related to the effects of the disease on the brain, limb muscles, respiratory system, heart and perhaps to altered levels of testosterone and insulin.
Congenital MMD1 threatens young lives, but children can improve
The most serious form of MMD1 — congenital-onset MMD1 — makes itself known at birth.
 Babies born with congenital MMD1 have very weak muscles and lack of muscle tone (hypotonia). They appear floppy and have trouble breathing, sucking and swallowing.
Babies born with congenital MMD1 have very weak muscles and lack of muscle tone (hypotonia). They appear floppy and have trouble breathing, sucking and swallowing.
There are always abnormalities in cognitive function, although intelligence can be in the normal range.
Speech and hearing difficulties often occur, and weakness of the eye muscles can cause vision problems. (Cataracts aren’t a feature of congenital MMD during early childhood, but develop later, as they do in adult-onset MMD.)
In the past, many babies with congenital-onset MMD didn’t survive, and death in the early months isn’t uncommon even now. But with modern newborn intensive care units, these babies have a much better chance of survival.
With much support during the early years, children with congenital-onset MMD can thrive. Their muscles become stronger as they grow, and they usually walk and reach major motor milestones.
Although some cognitive difficulties might not improve, most children can learn when given the right tools and environment. They often need help developing alternate means of communicating, so they can overcome the speech and writing difficulties caused by mouth, tongue and hand weakness.
Unfortunately, as children with congenital MMD approach adulthood, they start to develop the progressive features associated with the adult form of the disease.
Juvenile-onset MMD1 is 'somewhere in between' congenital and adult-onset forms
Some children develop MMD1 during their preschool or school years, although they aren’t born with symptoms of the disease. These children do not have the severe medical problems of babies with congenital-onset MMD1. However, cognitive difficulties can be serious, presenting challenges in school and social life.
The cognitive aspects of juvenile-onset MMD1 may overshadow the muscle aspects of the disease, although eventually, the muscle weakness will become apparent as well. The heart can be affected and needs attention.
Children with juvenile-onset MMD1 need lots of medical, educational and psychosocial support as they move toward adulthood.
DNA expansion on chromosome 19 underlies MMD1
In 1992, multiple research teams, some sponsored by MDA, simultaneously identified what’s now known to be the root cause of all forms of MMD1 — an expanded area of DNA on chromosome 19.
The area contains hundreds to thousands of repeated sequences of cytosine, thymine and guanine — or CTG — three DNA components. These CTG repeats are normal components of a gene known as DMPK, but the usual number of repeats ranges from three to 37. In people with MMD1, the CTG sequence is repeated at least 50 times.
 At the lower end of the expansion range (about 50 to 80 repeats), symptoms may be very mild or nonexistent. Cataracts are common, but they often don’t lead to an MMD diagnosis.
At the lower end of the expansion range (about 50 to 80 repeats), symptoms may be very mild or nonexistent. Cataracts are common, but they often don’t lead to an MMD diagnosis.
In general (but not reliably), the more prounounced the symptoms and the earlier the disease begins, the more repeats there are. The “classic” disease range (for adult-onset MMD1) is often between 100 and 500 repeats. Children born with the congenital-onset form can have thousands of CTG repeats.
And, in general (but again, not always), the number of repeats expands when the chromosome 19 gene mutation is passed from parent to child, and this expansion correlates with more severe symptoms and earlier disease onset. (The phenomenon of a disease getting worse and starting earlier as it’s passed from generation to generation is called anticipation.)
The congenital-onset form of the disease is found almost exclusively when the transmitting parent is the mother.
It should be noted that the number of repeats changes with time (generally expanding with age) in the tissues of an affected person, and it can differ in different tissues, such as blood cells and muscle cells. Therefore, the number of repeats depends on which tissue is sampled and the age of the person at the time the sample is taken. This means repeat number is not necessarily an accurate predictor of disease onset or severity; its main use is to unequivocally determine which individuals have MMD1 and which do not.
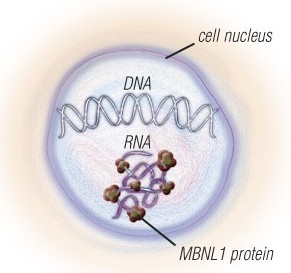
The nerve and muscle cells of people with MMD1 and MMD2 have expanded sections of DNA, which are converted to expanded sections of RNA.
The RNA traps a protein called MBNL1 and has other toxic effects on the cells. Several experimental therapies that target the expanded RNA are in the pipeline.
Expanded DNA on chromosome 3 causes MMD2
During the 1990s, genetic testing for MMD became widespread, and a curious phenomenon was uncovered: Some people who appeared to have the disease did not have the expected CTG repeat expansion on chromosome 19.
 Neurologist and neurophysiologist John Day was at the University of Minnesota at the time and was seeing patients in the MDA clinic in Minneapolis. (He’s now in California, heading Stanford University’s neuromuscular disease program and MDA clinic.)
Neurologist and neurophysiologist John Day was at the University of Minnesota at the time and was seeing patients in the MDA clinic in Minneapolis. (He’s now in California, heading Stanford University’s neuromuscular disease program and MDA clinic.)
"When I came to Minnesota in 1992, I worked with a family my predecessor had already identified as having myotonic dystrophy," Day recalls. "The genetic test had just become available for what turned out to be myotonic dystrophy type 1. So I said, 'That’s great. Why don’t we send off the genetic test just to confirm things?' It came back negative, which I thought was curious.
"Then within a week or two, a woman came into the clinic from a different family. She was pregnant, and she said her father had myotonic dystrophy and she wanted to know if she carried the genetic mutation."
Day asked her if she ever had problems releasing her grip on objects, and she said, "You mean like when I was driving down here today, and I couldn’t let go of the steering wheel?"
Strongly suspecting MMD, Day sent her blood to be tested — and her test also came back negative. "I thought, 'That’s good, but what the heck is going on?'" Day says. "I had thought she was affected. She had weakness and grip myotonia, and her father clearly had myotonic dystrophy. But here was a second family that was genetically negative."
By 1993, Day had seen two or three more families who appeared to have MMD but whose DNA tests were negative, and doctors in Germany were reporting similar families.
 Day talked to Laura Ranum, a neurogeneticist at the University of Minnesota who, with Day and neurologist Ken Ricker in Germany, would soon become involved in tracking down the origins of what eventually would be called type 2 myotonic dystrophy (MMD2). (Ranum, who has received several MDA research grants to study MMD, has since relocated to the University of Florida.)
Day talked to Laura Ranum, a neurogeneticist at the University of Minnesota who, with Day and neurologist Ken Ricker in Germany, would soon become involved in tracking down the origins of what eventually would be called type 2 myotonic dystrophy (MMD2). (Ranum, who has received several MDA research grants to study MMD, has since relocated to the University of Florida.)
In 2001, a team that included Ranum and Day and was funded in part by MDA announced the surprising findings: MMD could be caused by either of two genetic mutations — the CTG repeat expansion on chromosome 19 that had been identified in 1992, or a newly identified CCTG repeat expansion on chromosome 3 in a gene called ZNF9.
The number of CCTG (cytosine, cytosine, thymine, guanine) repeats in an MMD2-causing expansion averages 5,000, while the normal number is less than 50. The average repeat expansion in MMD2 is therefore much larger than in MMD1.
MMD2 is, in general, a somewhat milder condition than MMD1, although it shares features with that disorder. Both are characterized by myotonia, cataracts, defects in conduction of cardiac impulses, and an initial pattern of weakness that affects the muscles that flex the neck and fingers.
MMD2 has been called proximal myotonic myopathy, or PROMM, because the initial symptoms often involve difficulty arising from the floor or a low chair because of progressive weakness of hip musculature — the proximal muscles. This is in contrast to MMD1, where weakness of distal muscles in the hands is often an early symptom.
There are differences in the pattern of weakness between MMD1 and MMD2, but current nomenclature emphasizes the similarities in the two diseases, Day says.
One difference between the forms of MMD is that no congenital form of MMD2 has been seen so far. "I think the safest thing to say at this point is there’s no evidence of a congenital form" in MMD2, Day says.
And MMD2 does not seem to appear at an earlier age or to increase in severity with each generation, as MMD1 does.
Mild or moderate cognitive impairment can occur in both forms of the disease, Day says. Major cognitive impairment, such as that sometimes seen in congenital-onset MMD1, has been reported in some individuals with MMD2, but, Day notes, it could be merely a coincidence and not directly related to the MMD2 genetic mutation.
Tiredness and fatigue occur in MMD2, but the extreme sleepiness that can occur in MMD1 does not seem to be common in MMD2. “Clearly, more research is necessary to clarify this point,” says Day.
Similarly, in MMD2, Day says, cardiac problems are present at a higher rate than in the general population, and they can be very severe and life-threatening in some individuals, but they’re not as "predictably present" as in MMD1.
Families with MMD1 and MMD2 come to doctors’ attention for different reasons, Day notes. "What brings all too many people with MMD1 to clinical attention is that they have a child in the family who has congenital-onset disease," Day says. "Unfortunately, very few people with adult-onset or mild MMD1 receive an accurate diagnosis unless their child, niece or nephew is identified as having congenital-onset or juvenile MMD1."
Because there is no known congenital form of MMD2, he says, many with this disease come to clinical attention only after they reach 30 or 40 years of age and start having proximal leg weakness. (Adult MMD2 patients are often misdiagnosed as having an inflammatory myopathy or another muscle disorder, and the correct diagnosis can be missed for years.)
MMD2 may be MMD1 minus the 'developmental' aspects
Day’s way of looking at the two forms of the disease is that MMD1 has a “variable but potentially profound developmental defect” that can produce problems in infancy or childhood, while MMD2 does not appear to have this developmental component.
The scope and degree of developmental abnormalities (those that start early in life) vary in MMD1, Day notes. There can be weakness of all muscles, including those involved in breathing and swallowing; or weakness primarily of the muscles of the head and face, changing their shape and affecting speech; and there can be effects on brain development, potentially causing cognitive or behavioral abnormalities.
Day says, "These developmental problems can be incredibly profound and come to light at birth; or they can be moderate and show up during childhood; or they can be minimal and only be identified incidentally on examination of affected adults."
He says some people with MMD1 who come to the attention of doctors as adults may not have had any medical problems until recently, but close examination may uncover a thin face and high-arched palate, indicating muscle weakness of these areas during early life. And careful testing may reveal cognitive features that reflect developmental aspects of the disease.
"A major distinction between the two forms of myotonic dystrophy is that MMD1 has this developmental component, which can be variably severe from person to person, but which we do not recognize at all in MMD2," Day says. But, he says, in either form of MMD, there is a later-onset, progressive component starting in or near adulthood that affects skeletal muscles, the heart, brain, eyes and other organs in a very similar way.
"An interesting and currently unresolved question is why the CTG expansion in MMD1 can cause both the early developmental and later progressive problems, but the CCTG expansion in MMD2 either does not cause any developmental changes or causes them to such a minimal extent that they escape routine detection."
Where the research is headed
Several MDA grantees are working on strategies to block or destroy the repeat expansions in MMD1 and MMD2. Small molecules, such as pentamidine, and a strategy known as antisense are in development.
For videos covering some of the main research goals in MMD, see Myotonic Muscular Dystrophy Research Update for Families on the MDA website. Look for more about MMD research in the July-September 2012 issue of Quest.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
