
Connecting the Dots

Research shows congenital MDs have roots outside cells
Just a few years ago, almost nothing could be said to parents like those in “Families Left with Questions,” other than that their child had a congenital (present at or near birth) form of muscular dystrophy.
Andrew Loewi of Denver compares research on his daughter Samantha’s congenital MD (CMD) to Duchenne MD, for which the gene was identified in 1986. “What was so discouraging, so exasperating, for us, was that without even knowing what gene was involved, it seemed we were so far behind the eight ball that we’d never catch up.”
Doctors and scientists shared their frustrations throughout the 20th century. But several answers have emerged recently.
'Too much to hope for'
 In 1903, London physician Frederick Batten described a muscle disease of congenital onset, characterized by a lack of muscle power and tone, contracted joints and normal intelligence.
In 1903, London physician Frederick Batten described a muscle disease of congenital onset, characterized by a lack of muscle power and tone, contracted joints and normal intelligence.
Over the next several decades, doctors described sets of symptoms (syndromes) that they hoped represented unique genetic muscle diseases of congenital onset.
German physician Otto Ullrich reported in 1930 on children with a congenital atonic (without tone), sclerotic (with hardened scar tissue) muscle condition, curiously also characterized by highly flexible fingers and only mild weakness of the large muscles.
Then in Japan, in 1960, Yukio Fukuyama described a form of CMD that attacked not only muscles but the central nervous system, with severe mental retardation a frequent finding. About the same time, doctors in Finland called a similar syndrome muscle-eye-brain disease, and elsewhere another such set of symptoms was called Walker-Warburg syndrome. (North American physician Arthur Walker and Danish ophthalmologist Mette Warburg are credited with identifying this last.)
But as late as 1986, writing in the second edition of A Clinician’s View of Neuromuscular Diseases, Michael Brooke, now a professor emeritus at the University of Edmonton in Alberta, Canada, admitted he remained frustrated by the plethora of descriptions of CMDs and the blurring of the lines between them.
“Just when I think I have the entities clearly in mind, a new patient comes along with features common to more than one of these entities,” he wrote. “What is badly needed is some irrefutable biochemical test which will separate the diseases one from the other. This, of course, is too much to hope for ...”
Brooke, however, wouldn’t have to wait quite as long as he feared for progress on understanding congenital MDs at genetic and molecular levels.
Mapping the membrane
The year that Brooke published his clinical textbook, MDA-supported researchers working in the laboratory of Louis Kunkel at Children’s Hospital in Boston identified mutations in a large, X chromosome gene for a protein that would come to be known as dystrophin as the underlying cause of Duchenne muscular dystrophy, a common and severe childhood form of MD.
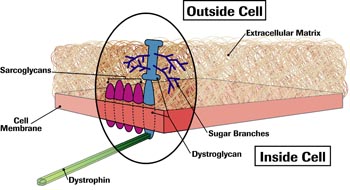
Dystrophin, it was soon discovered, lies just under the muscle cell membrane, a thin sac that encloses muscle cells and determines which substances can enter and leave them.
As early as 1989, researchers in the laboratory of Kevin Campbell, an MDA-funded protein chemist at the University of Iowa, had found that dystrophin is attached to the membrane — not directly, but through another compound they called a glycoprotein, because it was combined with sugar (glyco) molecules.
By the mid-1990s, Campbell and others had described four other membrane-embedded proteins, which came to be known as sarcoglycans, and a two-part protein that Campbell named dystroglycan. All these proteins had sugar branches protruding from their surfaces, and dystroglycan had one section stuck in the membrane and another protruding into the gluelike substance outside the cell — the extracellular matrix.
It was also becoming clear that the proteins in this membrane complex (cluster) — each of which is made by a different gene — closely interacted with one another, and that the loss of any of them affected the ability of this dystrophin-glycoprotein complex (DGC) to assemble. As a result, the muscle cell membrane was weakened and its function impaired. (Loss of any of the four sarcoglycans leads to limb-girdle MD.)
Moving on up — and out
Beyond the cell membrane, in the extracellular matrix, lie proteins called laminins (layers) and collagens (glue makers), among other things. Also beyond the cell membrane is another, tougher membrane, itself made of two layers, the basal and the reticular (netlike) lamina. Laminin 2 (merosin) sticks up out of the DGC and into the basal lamina.
In 1993, Campbell and another MDA grantee, James Ervasti, showed that dystroglycan, the sugary protein that protruded from the muscle cell membrane, interacted closely with the muscle form of laminin outside the cell. (At the time, the laminin found in muscle tissue was widely known as merosin, though it was soon to be rechristened laminin 2.)
At the time, French physician Fernando Tome was looking at biopsy samples from children with CMD and normal brain function. He noted that merosin was absent in 13 of them.
Tome visited Campbell’s lab just after the finding that dystroglycan was bound to merosin, and he wondered if merosin deficiency could be the direct cause of the CMD in his patients.
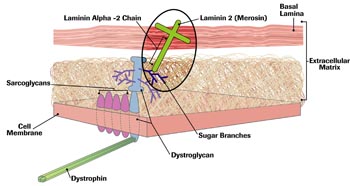 In 1995, Tome, as part of a group based at the Pitie-Salpetriere Hospital in Paris, published a paper showing that one of three of merosin’s protein chains, made from a gene on chromosome 6, could, when flawed, cause CMD. The protein, the alpha-2 chain, is in the center of a three-chain structure. The other chains, from two different genes, were intact in Tome’s patients, but without the center strand, they were unanchored and useless to the cell.
In 1995, Tome, as part of a group based at the Pitie-Salpetriere Hospital in Paris, published a paper showing that one of three of merosin’s protein chains, made from a gene on chromosome 6, could, when flawed, cause CMD. The protein, the alpha-2 chain, is in the center of a three-chain structure. The other chains, from two different genes, were intact in Tome’s patients, but without the center strand, they were unanchored and useless to the cell.
The first dot — a specific protein defect linked to a congenital MD — had been drawn, and physicians began to describe congenital MD as either merosin-deficient or merosin-positive. The classification didn’t help much in predicting the course of the disease and turned out to be confusing because merosin can be deficient for reasons other than the chromosome 6 gene mutations. But it was a start.
The collagen connection
As the 20th century came to an end, most investigators had forgotten about Otto Ullrich’s 1930 paper. Ullrich had described a congenital-onset muscular dystrophy, with normal intelligence, a striking combination of contractures in some joints and laxity in others, reduced bone density, and skin that contained rough patches with raised bumps resembling sandpaper on the thighs, shins and upper arms, with velvety, smooth skin on the soles and palms.
 In the early 1990s, Carsten Bonnemann, born and trained in pediatrics in Germany, came to the United States to undertake postgraduate training in child neurology at Massachusetts General Hospital and Children’s Hospital in Boston, and later a program in genetics in Kunkel’s laboratory at Children’s.
In the early 1990s, Carsten Bonnemann, born and trained in pediatrics in Germany, came to the United States to undertake postgraduate training in child neurology at Massachusetts General Hospital and Children’s Hospital in Boston, and later a program in genetics in Kunkel’s laboratory at Children’s.
“Kunkel set me onto the dystrophin-associated glycoproteins,” Bonnemann recalls, “because, at the time, that was just coming up. And I identified the beta-sarcoglycan gene.
“The sarcoglycans stick out through the cell membrane, and unlike dystroglycan, they don’t have a ligand [attaching molecule] binding to them. They stick out and look like orphans that should be binding to something in the matrix. That’s how I got interested in the extracellular matrix.”
Bonnemann and other researchers are searching for a link from the sarcoglycans to the extracellular matrix that may be parallel to the dystroglycan-merosin link.
In 2000, back in Germany, he began seeing patients with the syndrome Ullrich had described long ago. Because of their joint laxity, they reminded him of a connective tissue disorder called Ehlers-Danlos syndrome and also of a family he’d seen in Boston. Members of the Boston family had a known defect in one of the collagen proteins, with severe joint abnormalities and mild muscle problems.
Bonnemann wondered whether the patients with joint and muscle symptoms described by Ullrich could also have a collagen defect.
 Two years later, back in the States, this time at Children’s Hospital of Philadelphia, Bonnemann saw other children with congenital-onset weakness and marked contractures in some joints, combined with joint laxity elsewhere. Many had gone without a diagnosis for some time.
Two years later, back in the States, this time at Children’s Hospital of Philadelphia, Bonnemann saw other children with congenital-onset weakness and marked contractures in some joints, combined with joint laxity elsewhere. Many had gone without a diagnosis for some time.
“They often had an odyssey through genetics clinics and connective tissue clinics,” Bonnemann says, “with nobody really coming up with a diagnosis.” Bonnemann recognized them as having Ullrich congenital MD.
“Once you’ve seen these patients, it’s recognizable as a unique disease,” he says. “It’s probably one of the more common forms of congenital muscular dystrophy — in this country probably more common than merosin deficiency.” Based on the appearance of the patients, Bonnemann suspected collagen was involved — a hunch that turned out to be correct.
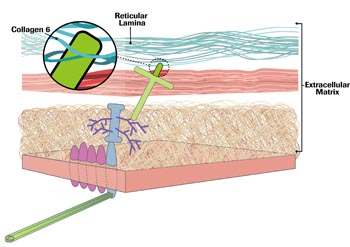
Mutations that affect collagen 6, one of more than 25 collagen types, were found to cause Ullrich CMD by researchers in Italy in 2001, investigators in France in 2002, and a Philadelphia-based team that included Bonnemann in 2003.
Like laminin 2, collagen 6 is made up of three chains, alpha-1, 2 and 3, each arising from a different gene. Mutations in any of the three can disrupt the triple-stranded molecule and lead to connective tissue, muscle, skin and bone abnormalities. Depending on the type of genetic mutation (but not on the gene), the resulting disorder can be Ullrich CMD or Bethlem myopathy, a less severe version of the same disorder, with a somewhat later onset.
Collagen 6, Bonnemann says, is attached to the basal lamina. But, “It seems to be reaching into the basal lamina, coming from the outside, from further outside the cell.”
A second dot was on the CMD drawing board, and lines could be drawn connecting it to collagen 6. Of equal importance, lines now connected two forms of CMD to the extracellular matrix, and to each other.
The sugar connection
 At the end of the 20th century, a lot of progress had been made in classifying the congenital MDs, but one glaring question remained: Why did some CMDs, such as Fukuyama CMD, muscle-eye-brain disease (MEB) and Walker-Warburg syndrome (WWS), involve the brain and eyes, while others, such as the collagen-deficient Ullrich CMD and merosin-deficient CMD due to alpha-2 chain mutations, apparently didn’t?
At the end of the 20th century, a lot of progress had been made in classifying the congenital MDs, but one glaring question remained: Why did some CMDs, such as Fukuyama CMD, muscle-eye-brain disease (MEB) and Walker-Warburg syndrome (WWS), involve the brain and eyes, while others, such as the collagen-deficient Ullrich CMD and merosin-deficient CMD due to alpha-2 chain mutations, apparently didn’t?
(Some forms of merosin-deficient CMD lack merosin as a secondary effect of the disruption of another protein. Unless that protein is known, the course of these merosin-deficient CMDs is uncertain.)
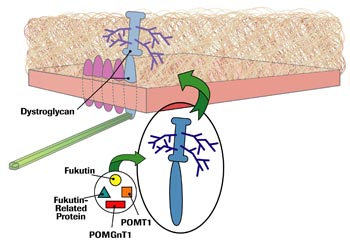
In 1998, Japanese researchers had identified a chromosome 9 gene for a protein they dubbed fukutin as the cause of Fukuyama CMD.
Then, in 2001, researchers at Hammersmith Hospital in London described patients with a severe CMD without brain involvement, and found that their disease was due to mutations in a chromosome 19 gene for a protein that closely resembled, but wasn’t, fukutin. They called it fukutin-related protein (FKRP).
They also noticed something else. Biopsies of these patients showed that laminin 2 was missing from its usual place protruding from dystroglycan, and that the part of dystroglycan that sticks out of the membrane didn’t look normal. When the sample was stained, dystroglycan showed up only faintly, and other tests showed it was markedly under its usual molecular weight.
The investigators, led by Francesco Muntoni, added a third dot to the CMD picture by proposing that fukutin-related protein might be involved in the sugar coating — glycosylation — of dystroglycan, and that its absence might result in a denuded dystroglycan incapable of sticking to laminin 2.
Scientists also found that fukutin was involved in glycosylation. Glycosylation is a common process in cells, involving a variety of enzymes, proteins that bring other compounds together or break them apart.
In 2001, researchers in Yokohama, Japan, described something that got everyone’s attention: a rare type of protein glycosylation operating in the nervous system and in muscles, catalyzed by the enzyme POMGnT1. They identified six different mutations in the gene for POMGnT1, each of which led to MEB, a CMD that Finnish investigators had described decades earlier.
Clearly, inadequate glycosylation of dystroglycan was the culprit in more than one form of CMD. Dots number 4, 5 and 6, closely connected to each other, to the DGC and to the extracellular matrix, could now be added to the enlarging diagram of this group of congenital disorders.
More evidence arrived the following year, when a group based at University Medical Centre in Nijmegen, the Netherlands, identified mutations in a gene for a glycosylation enzyme called POMT1 as the cause of Walker-Warburg syndrome in six people. POMT1’s function, as its name implies, is similar to that of POMGnT1, but it’s a different enzyme, arising from a different gene. Make that dot number 7.
A sugar-coated dystroglycan, it seemed, was necessary to brain and eye development as well as to muscles, so it wasn’t surprising that Fukuyama CMD, WWS and MEB affected all three systems. What remained puzzling was that FKRP-deficient CMD, also a glycosylation disorder, didn’t.
Research in the last couple of years has cleared up this mystery, even though it has ironically made predicting the course and severity of an individual case of CMD more difficult.
The research has shown that it isn’t the particular gene or glycosylation protein that matters when it comes to brain involvement, but the nature of the gene defect, or mutation. In 2004, for example, it became clear that some mutations in the FKRP gene can lead to a disease that’s indistinguishable from MEB and WWS, while other mutations affect only muscle tissue.
Creating more connections
 Of course, the question on the minds of parents and doctors is still: What can be done to treat congenital MDs?
Of course, the question on the minds of parents and doctors is still: What can be done to treat congenital MDs?
Right now, the available treatments are supportive. Physical therapy, spine-straightening and contracture-releasing surgery, good respiratory care, including assisted ventilation in some cases, are the mainstays.
Life span, Bonnemann says, is a “moving target” in Ullrich CMD.
“If you look in the old literature, the life span was into the second decade, and then they would die from respiratory complications,” he says. “But since we are now much better in getting on top of the pulmonary complications, the life span remains to be seen.
“We have one patient here who’s now 23, with a really severe form of Ullrich CMD. He uses a wheelchair and has many contractures, but he’s stable, with nighttime BiPAP [noninvasive assisted ventilation]. He has his own car and is an engineering major at Villanova College here.”
Research advances could make the future even brighter. In 2004, Kevin Campbell’s group added a glycosyltransferase that goes by the acronym LARGE to cells taken from people with several glycosylation-deficient forms of CMD, and found that sugaring of dystroglycan, as well as integrity of its connections, were restored.
And just last year, MDA grantees Chunping Qiao and Xiao Xiao at the University of Pittsburgh found that mice missing the laminin alpha-2 chain were substantially improved by the addition of genes for a protein called agrin, which may in part compensate for the absence of laminin 2. They used the agrin gene in a miniaturized form, because it's technically easier to work with than a laminin gene and because agrin is less likely to be rejected by the immune system where the laminin has been missing from birth.
Families Left with Questions
Samantha
Shortly after Denver couple Pat and Andrew Loewi adopted Samantha in 1989, they began to suspect that something was wrong.
Pat had a daughter from an earlier marriage, and she knew this baby wasn’t developing the same way Kim had. “She never crawled and didn’t move much, didn’t roll much, and yet she walked, with help, at 9 months,” Pat says.
In May 1991, the Loewis took 18-month-old Samantha to Children’s Hospital in Denver, where a physical exam immediately made the doctor suspect she had muscular dystrophy. A month later, a muscle biopsy confirmed the diagnosis but added little more, other than that the dystrophy wasn’t Duchenne.
Kendle
Born in 2002, Kendle is the first child of Robbie and Kristy Haught, in Washington, Pa., a suburb of Pittsburgh.
“When she was born, she was really floppy, really loose,” Kristy says. “At 2 months, I was concerned something was wrong, because she wasn’t holding her head up at all.” At 4 months, Kendle was admitted to Children’s Hospital of Pittsburgh for six days of tests; only a muscle biopsy provided any answers.
The biopsy showed muscular dystrophy, which provided relief to the Haughts, who had been reading about type 1 spinal muscular atrophy, a condition that often takes the lives of young children. Kendle’s disease, the doctors said, had a better prognosis than SMA, but they were uncertain what the future would hold.
Robyn
In San Jose, Calif., four years ago, Aarica and Oscar Gutierrez were asking some of the same questions about their baby, Robyn. Doctors said Robyn had low muscle tone (hypotonia). At home, Robyn was floppy, and her joints seemed very mobile, with fingers that bent back all the way and feet that bent back to her shins. On each shin was a patch of rough skin that looked to her parents like goose bumps.
She sat at 7 months and pulled herself to a standing position at 13 months. Her cognitive development, speech and vision were normal.
Her mother recalls, “At a year old, we were actually starting to get excited by the fact that she could pull up and could do some things, and I was really believing that she was just hypotonic, like the doctors said. Everything I read said that usually, by the time they’re 2 years old or a little past that, they’ll be fine.
“But after she hit the 18-month mark, we knew that wasn’t going to be the case.”
As she grew and gained weight, Robyn lost the ability to sit on her own and to pull up using the coffee table. She walked, but with bent knees, an awkward gait and frequent falls.
Some Answers
Samantha
 Samantha Loewi now has a specific, DNA-based diagnosis, but it took 15 years to arrive at it.
Samantha Loewi now has a specific, DNA-based diagnosis, but it took 15 years to arrive at it.
Now 16 and a sophomore in high school, she takes honors classes and plays the soprano saxophone and bass guitar. She has Ullrich CMD, with a mutation in a collagen 6 gene.
Her father, Andy Loewi, who describes her as “happy, well adjusted, fun and spunky,” says she’s “dealt with the rather immense challenges that confront her with amazing grace, courage and good humor.” He’s much encouraged by the specific diagnosis they now have.
“Knowing exactly what the gene defect is gives us so much hope that she will not have to wait to really see an effective gene therapy or some other treatment that will be effective in ameliorating or even curing the disease,” Andy says.
Samantha started using a power chair when she was 10, and her condition hasn’t changed much since then. “We’ve figured out a way for me to do a lot of things,” she says. Next on her agenda are starting a band with some musician friends and learning to drive an adapted vehicle.
Kendle
For the Haughts, the diagnostic odyssey isn’t quite over. Before Kendle was 5 months old, she had a biopsy that showed merosin-deficient CMD, but further details await DNA testing. She’ll be 4 in May, and attends a preschool.
 Kendle is weak but can support herself sitting if she’s not too tired. Severe contractures tighten her hips, knees, ankles and elbows, and keep her from lying flat if she’s on her stomach and from standing, even in a stander.
Kendle is weak but can support herself sitting if she’s not too tired. Severe contractures tighten her hips, knees, ankles and elbows, and keep her from lying flat if she’s on her stomach and from standing, even in a stander.
She can’t straighten her arms, but her fingers are quite dexterous, allowing her to write, feed herself and use her computer. She began talking at a year, and at 3, her cognitive development was so advanced that her preschool recommended she be put in a class of 4- and 5-year-olds.
When she was 2, her physical therapist suggested that her high intelligence and maturity would make her a good candidate for a power chair. “She was always cognitively ahead,” Kristy says, but even so, she wasn’t sure about a 2-year-old driving a power chair.
But, she says, “They had one there, and they got it, and the minute Kendle touched the joystick it was like she’d been doing it her whole life.”
Kendle also has a weak cough, which puts her at risk for serious respiratory infections, and has recently acquired a CoughAssist, which pulls mucus out of the lungs with suction. “It’s not easy,” says Kristy, who tried it on herself, “and she doesn’t like it at all,” but her parents hope it will keep her free of pneumonia.
A rapidly progressing spinal curvature (scoliosis) concerns her doctors, who say she’ll probably have to have surgery to correct it before long.
Kristy keeps in touch with other parents via Internet chats. Kendle hasn’t needed assisted ventilation or feeding tubes, and she has excellent cardiac function, good hearing and vision, finger dexterity and high intelligence as well.
“I feel we’ve been really fortunate,” Kristy says.
Robyn
Robyn Gutierrez, who will turn 4 in May, has Ullrich CMD, with a mutation in the alpha-3 chain of collagen 6. The Gutierrezes got her diagnosis last year after locating Carsten Bonnemann through an Internet search and sending a sample of Robyn’s muscle tissue to him. (They’re now seeing pediatric neurologist Jonathan Strober, director of the MDA clinic at the University of California at San Francisco Children’s Hospital.)
 She’s in a special preschool program that Head Start operates in California.
She’s in a special preschool program that Head Start operates in California.
“She’s very bright,” says her mother, Aarica. “Her speech is very clear and precise, and she has no problem letting you know what she wants. She speaks in full paragraphs.
“Physically, she’s still pretty much the same as when she was 18 months old. She hasn’t gotten any worse, but she hasn’t gotten any stronger or better. I think she can do a few more things, because with awareness of her limitations, she tries to compensate. She’s very good at compensating.” Even so, she falls often, and now wears a light, foam helmet to protect her head when she’s in school. She uses a walker when she’s outside on the playground.
“Robyn has more mobility than most UCMD children whose parents I speak with,” Aarica says. “But she’s only allowed to walk around independently at home in the carpeted playroom area. The full family room and the back of the house are pretty much her domain. After about 20 to 30 minutes, somebody has to be with her, because her legs get tired, and she tends to fall. We don’t have any area rugs in our house, because even those are hard for her to step up over.”
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
