
Matters of the Heart: Cardiac Problems in Emery-Dreifuss MD
Jason Adamo’s life isn’t markedly different from that of any other teenager. The 17-year-old high school junior has a part-time job as a cook at a restaurant near his home in Port Charlotte, Fla., and enjoys flying radio-controlled model helicopters in his spare time.
But had it not been for the alertness and persistence of his mother, Katherine, an intensive care nurse, things could have taken a tragic turn not long ago.
 Jason, like many teenage boys, isn’t much of a talker, so when he began complaining of severe belly pain last spring, his mother took it seriously. “We thought it might have been appendicitis,” she says, “because he had complained of it for a couple of days, and it started moving to the right.”
Jason, like many teenage boys, isn’t much of a talker, so when he began complaining of severe belly pain last spring, his mother took it seriously. “We thought it might have been appendicitis,” she says, “because he had complained of it for a couple of days, and it started moving to the right.”
In the emergency room that evening, Jason’s pulse rate was found to be 45 beats a minute. (A normal heart rate is between 60 and 100.)
The doctor wasn’t overly concerned, since athletic young men can have hearts that pump so efficiently that their rate can be quite low.
“Does he play sports?” the ER physician asked.
Adamo replied, “No, he does a lot of models. He doesn’t play football or anything. He tried when he was younger, but he had so many problems with his legs, he gave that up. He felt he couldn’t, that he wasn’t athletic.”
In fact, a couple of years earlier, Jason had started walking on his toes, and doctors had recommended surgery to lengthen his Achilles tendons, which run up the backs of the heels.
“The doctor thought his legs grew too fast, and the Achilles tendons weren’t long enough for all that growth,” Adamo recalls. The surgery, which required prolonged casting and bracing of Jason’s legs, had been successful. But afterwards, the family had noticed something else: Jason’s elbows were also getting tight — developing contractures — and he could no longer fully straighten his arms.
When Adamo had asked doctors if there could be a correlation with his leg problem, they said they didn’t think so.
Now, in the ER, she recalled another unpleasant fact. Her father had died when she was 13 and he was in his 30s, of a heart problem. He’d also had a massive stroke at age 24 and more strokes after that, spending the last years of his life severely disabled.
A diagnosis at last
Jason was referred to a cardiac electrophysiologist, a physician who specializes in the impulses that travel through the heart and cause it to beat regularly most of the time.
 The specialist ordered thorough and specific testing, and the results weren’t exactly comforting. The upper chambers of Jason’s heart (the atria) were contracting at an almost unbelievable 380 beats per minute, while at the same time the lower chambers (ventricles), where the main pumping action is, dropped into the 30s while Jason was sleeping. Signals from the atria weren’t getting through to the ventricles, where the heart’s main pumping action takes place.
The specialist ordered thorough and specific testing, and the results weren’t exactly comforting. The upper chambers of Jason’s heart (the atria) were contracting at an almost unbelievable 380 beats per minute, while at the same time the lower chambers (ventricles), where the main pumping action is, dropped into the 30s while Jason was sleeping. Signals from the atria weren’t getting through to the ventricles, where the heart’s main pumping action takes place.
A heart rate in the 30s is dangerous. “That’s how some of these kids end up having these sudden deaths,” Katherine Adamo says. “They either drop from a stroke at a very young age, or they just have sudden death.”
Fortunately, Jason’s story has a happy ending. He and his parents ended up at the MDA clinic at St. Joseph’s Children’s Hospital of Tampa, Fla., with pediatric neurologist Raymond Fernandez, the clinic director.
“I’m sitting there with my husband [Louis], and I’m thinking, ‘You see, there’s nothing wrong with him,’ because when you think of muscular dystrophy, you always think of muscle weakness; and Jason, from the time he was born, I always thought his muscle tone was a lot tighter than my other two children.
“But once Dr. Fernandez looked at Jason and heard the history of his Achilles tendons, his difficulties with athletics and his heart abnormality, he said, ‘OK, this is what I think we’re dealing with,’ and recommended DNA testing for Emery-Dreifuss muscular dystrophy.
“We were kind of floored,” Adamo remembers, “and then we did the DNA testing, and it all came back positive.”
Jason’s heart problem was ultimately treated by the implantation of an electronic pacemaker, which, he says, he doesn’t even remember is there. He’s back to all his usual activities and plans to be a fire fighter and emergency medical technician after high school.
Preop tests save a life
Just a few years before Jason Adamo’s trip to the emergency room in Florida, José Cereceres, then a 40-year-old accountant at White Sands Missile Range near Las Cruces, N.M., was undergoing routine preoperative tests before having his gallbladder removed when he and his doctors got a surprise.
“They said my heart couldn’t take the surgery,” Cereceres says. “I was told I had electrical short circuits in the heart. They put in a pacemaker, and then a little over a month later, they took out the gallbladder. That’s how I got the pacemaker, because of that surgery.”
Unlike Jason Adamo, Cereceres knew at the time of his tests that he had Emery-Dreifuss MD, but he hadn’t known for long, and he wasn’t aware of any heart problems.
As a child, he fell down a lot and walked slowly. By his teens, he was starting to walk on tiptoe, with tight heel tendons. The MDA clinic in Albuquerque helped him find a surgeon to relieve the Achilles tendon shortening, and referred him for physical therapy, but no one was sure about the diagnosis.
Then, a few years ago, Cereceres, who’s now 43, went to the MDA clinic in El Paso, Texas, where he was referred for genetic testing and finally learned that he had X-linked EDMD.
Unlike the Adamos, Cereceres doesn’t know of any family history of either heart or muscle problems. His running and walking have gradually slowed, and his falls have become more frequent.
“Right now, I think my main concerns are balance and trying not to fall,” he says. “My arms are constricted. I can’t stretch them all the way out, and I find as I get older that my feet are becoming more tightened.”
But his heart problem is, for now at least, under control.
Two weeks after having the pacemaker inserted, Cereceres was back at work. “Everything’s fine so far,” he says, “although it’s kind of funny having something inside you with a wire that goes up to your heart. It’s amazing what they can do these days, the technology. I’m just grateful that I’m healthy enough to continue having a normal life.”
A 'benign' form of MD
Emery-Dreifuss MD is named for Alan Emery, a British physician and medical geneticist, and Fritz Dreifuss, a German-born neurologist. Emery is now chief scientific adviser for the European Neuromuscular Centre, based in Baarn, the Netherlands, and an emeritus professor at the University of Edinburgh in Scotland. Dreifuss, who died in 1997, trained in New Zealand and England and was a professor of neurology at the University of Virginia in Charlottesville.
In 1966, Emery and Dreifuss published a paper in the Journal of Neurology, Neurosurgery and Psychiatry describing “an unusual type of benign X-linked muscular dystrophy.” The word benignmeant it wasn’t very serious, at least in comparison to the fatal Duchenne MD that was already well recognized.
Describing the new dystrophy, which they had observed in a large, multigeneration family in Virginia, they wrote, “All the affected members have the following features in common: contractures of both elbows and shortening of the tendon Achilles dating from early childhood, mild pectus excavatum [depression of the breastbone], and absence of pseudohypertrophic calves [the enlarged calf muscles typical of Duchenne MD].... None are mentally retarded and in fact two are schoolteachers. Among the older affected males abnormal cardiac rhythms, nonspecific murmurs and sudden death have been observed.”
Writing in the June 2000 issue of Neuromuscular Disorders 34 years later, Emery noted that the disorder is characterized by early contractures, often before there’s any significant weakness; slowly progressive muscle wasting and weakness, starting in the upper arms and lower legs and progressing to the shoulder and hip muscles, although never becoming profound; and cardiac conduction (signal transmission) defects.
He could also note that the disease comes in two forms: the more common, X-linked recessive form, resulting from a mutation in an X chromosome gene for a protein called emerin (after Emery) and identified in 1994; and an autosomal (non-X-linked) form, arising from mutations in a gene on chromosome 1 for lamin A/C, a link identified in 1999.
“Cardiac involvement is the most serious and important aspect of the disease,” Emery wrote in 2000. The heart is a muscle, and the one most seriously affected in EDMD.
Effects on the heart
“In the past, people have defined Emery-Dreifuss as a not very severe form of muscular dystrophy, because the skeletal muscle involvement hasn’t been as severe [as in other types of MD]; but that’s not been true about the heart involvement,” says William Groh, a cardiac electrophysiologist and MDA research grantee at Indiana University in Indianapolis.
 “In Emery-Dreifuss MD, the early [cardiac] involvement is primarily arrhythmias [abnormal heart rhythms],” Groh says. “It’s very common for men with the X-linked recessive form, by their 20s and 30s, to show electrocardiographic abnormalities and have arrhythmias.” Female carriers of the X-linked form are also at risk for these problems, although typically later in life.
“In Emery-Dreifuss MD, the early [cardiac] involvement is primarily arrhythmias [abnormal heart rhythms],” Groh says. “It’s very common for men with the X-linked recessive form, by their 20s and 30s, to show electrocardiographic abnormalities and have arrhythmias.” Female carriers of the X-linked form are also at risk for these problems, although typically later in life.
The heart’s atria seem to be involved earlier than the ventricles. “The patients can be prone to atrial fibrillation and atrial flutter [very fast and ineffective atrial rates], and these patients can get something called permanent atrial standstill.”
The latter, Groh says, reflects such a severe scarring of the atria that electrical signals don’t move through them at all. “The atria just sit there.”
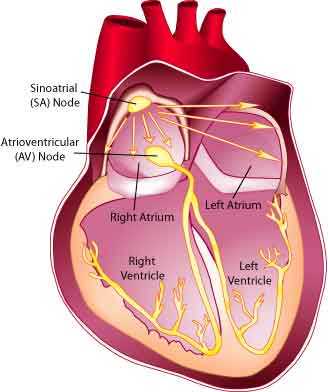
The heart’s normal pacing center is the sinoatrial (SA) node, located high in the right atrium. It sends signals to a major relay station, the atrioventricular (AV) node, which is located where the atria and ventricles meet. The impulse is then conducted through the rest of the fibers, causing the muscular contractions that pump blood around the body.
Typically, if the sinoatrial node isn’t working because of scar tissue, the atrioventricular node takes over, setting a heart rate of about 40 beats a minute. If the AV node is likewise nonfunctional, pacemakers lower down in the heart take over, usually dropping the heart rate to about 30 beats per minute.
The track followed by the electrical impulses is called the conduction system, and it seems particularly hard hit in EDMD.
“In the atrium, the myocytes [muscle cells] in the conduction system don’t look different from other cells,” Groh says. “It may be that that tissue is just so much more metabolically active that it fibroses [scars] more quickly.” Lower down in the heart, he says, these conducting cells do look a little different from the other cells. “Their job is to rapidly conduct electricity through the heart. It’s kind of like the freeway of the heart, with everything else being little highways coming off the freeway,” he says.
Some people may feel palpitations or a fluttering sensation in the chest or may be aware of fast or irregular heartbeats. Fainting spells may occur with a heart rate that’s too slow or too fast.
But sometimes the symptoms are vague. “We’ve had patients come in who say, ‘Boy, Doc, for the last three months, I just haven’t felt well,’ and we find out that in fact they have a conduction block and a heart rate of 30 beats a minute.”
Abdominal pain, such as Jason Adamo experienced, or nausea, can also occur. “When the heartbeat slows, the body tries to react to that by building adrenaline levels up, trying to get the heart rate back up, and that can lead to a lot of different symptoms,” Groh says.
Most patients will ultimately require pacemakers to regulate the heartbeat, he notes, although sometimes other treatments are tried first or added. People who have both fast and slow heart rhythm abnormalities may receive an implanted pacemaker-defibrillator, which not only paces the heart but can also convert a fast, irregular beat back to a normal rhythm.
Doctors may also prescribe drugs that lessen the heart’s workload, while drugs to prevent clots from forming (anticoagulants) are routinely prescribed for patients with atrial fibrillation.
Another treatment is radio frequency catheter ablation, a therapy in which low-voltage, high-frequency electricity is used to obliterate (ablate) an abnormal signaling pathway in the heart, via a thin tube (catheter) inserted through a blood vessel. If the main problem is atrial fibrillation or flutter, a cardiologist can ablate the AV node, preventing the dangerous signals from moving to the lower heart. After the AV node is ablated, the doctor inserts a permanent pacemaker.
Cell structure, gene activity in the crosshairs
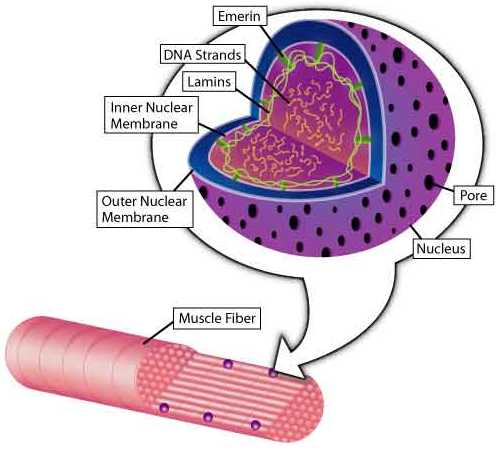
“The findings of emerin and lamin mutations in Emery-Dreifuss dystrophy were really complete surprises,” says Howard Worman, a physician and MDA grantee at Columbia University in New York, where he’s studying proteins that make up the nuclear envelope, a structure that surrounds each cell nucleus.
In the ‘90s, he says, scientists were "finding genes by their position. They got families with a disease, then they homed in on the genes, and then they found the mutations.” (Geneticists analyze sections of DNA from affected and unaffected family members until they find a key section that always correlates with having the disease.)
“When a research group in Italy found the emerin gene and the protein it instructs for, in 1994, it was until then unknown. About two years later, groups in Wales and Japan localized the protein to the nuclear envelope," Worman says.
“Lamins A and C were better known proteins. We had analyzed the gene’s structure back in 1993 because we were working on lamins. Then in 1999, scientists in France identified mutations in lamins A and C as causes of EDMD. The gene for these is called LMNA, or the lamin A/C gene, because it contains the code for both those proteins, depending on how the genetic message is processed.”
Emerin mutations underlie the X-linked form of the disease, and lamin mutations the chromosome 1 form.
“Clinically, they’re indistinguishable,” Worman says, “but the inheritance is different. Only males get the X-linked form, although some females develop cardiac abnormalities or weakness. In the chromosome 1 form, it’s males and females who get it.”
Recent studies have shown that the chromosome 1 form can be inherited in an autosomal dominant or, very rarely, a recessive pattern. Genetic testing is available for all forms of EDMD.
(X-linked disorders are usually carried by women, who have two X chromosomes, one of which is unaffected and therefore keeps the full disease from showing itself. Autosomal dominant disorders manifest themselves even when only one mutated gene is inherited, while autosomal recessive disorders require that a child inherit a mutation in the same gene from each parent.)
More questions about proteins
After the 1999 linkage of lamins A and C with EDMD, biologists got yet another surprise, when they found that different lamin mutations could cause either muscle disease, fat cell disease, peripheral nerve disease or premature aging.
 But crucial questions about how abnormalities in any of these proteins lead to EDMD or any other disease remain largely unanswered.
But crucial questions about how abnormalities in any of these proteins lead to EDMD or any other disease remain largely unanswered.
“One is that mutations in either emerin or lamin A/C may have some structural or support or mechanical effect on cells,” Worman says, and that they’re necessary for cellular integrity. That may explain why certain mutations cause muscle disease, because muscles are under mechanical stress more than other cells are.
“The other hypothesis — and these aren’t mutually exclusive — is that somehow the nuclear envelope is involved in regulating the activity of genes in specific tissues.”
How they might do that is so far unknown, but scientists are beginning to form hypotheses. “When the cell isn’t dividing, the chromosomes are there, but they’re spread out all over the nucleus,” Worman explains. “They’re not the structures you think of as chromosomes. They’re just strands of DNA lying every which way.” Nuclear envelope proteins and chromosomes might interact at this or other time points.
As to the search for EDMD’s molecular roots, Worman says, “Either it’s going to be something mechanically wrong with the cells, or there’s going to be abnormal gene activity that results from these mutations, or both. And the gene expression [activity] may be from genes that are important in muscle differentiation, maintenance, stability or regeneration.
“It’s funny that the heart seems to be always affected, while the skeletal muscle is to a more variable degree. I’d say it’s not really clear why,” he says, adding that the reasons why some muscles are more affected than others and why various tissues are affected by different lamin mutations are likewise unknown.
“There’s pretty good evidence that emerin and the lamins bind [stick] to each other,” Worman says. “Lamins A and C bind to emerin. You seem to need lamins A and C to retain emerin in the inner nuclear membrane, one of the layers of the envelope. In the absence of lamins A and C, emerin seems to diffuse out of the nuclear envelope. The two definitely stick together in cells, but the significance of the interaction isn’t well understood yet.”
Worman says mice bred with lamin mutations develop heart and skeletal muscle disease and should yield clues in the near future, as should mice lacking emerin that are now being developed.
From theory to therapy
As to treatment, Worman says gene therapy to insert an emerin gene isn’t impossible to think about, especially since the gene is relatively small and so far it appears that the problem is emerin deficiency, rather than emerin abnormality or toxicity.
“I don’t want people thinking that we’re going to inject an emerin gene into them and they’re going to be all better,” Worman says. “But it looks like there’s a loss of emerin, so theoretically gene therapy to replace emerin, or cell therapy with cells that contain emerin, could be tried.”
Lamin mutations, he says, are another story.
“There are some mutations where the lamin is probably not expressed [produced], and there are others where the mutant protein seems to hang around. I think one question that we may answer through work in cells and in mice is: Is it always just loss of the lamins A and C, or is it sometimes the presence of a mutated lamin A and C, that causes the problem? In the first case, theoretically, you could use gene therapy. In the second case, you would have to do something to get rid of the mutated protein.”
But there are other baskets for research eggs.
“If we can understand the mechanisms, if for instance the lamin mutations make certain genes turn on and off, you might find the pathways that are abnormally regulated and treat the problem with agents that compensate for those changes,” Worman speculates.
He adds that the most serious problem in EDMD is cardiomyopathy, and that today’s electronic devices are very effective.
“Today, with pacemakers and implantable defibrillators, people can live comfortably for a long time.”
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
