Amyotrophic Lateral Sclerosis (ALS)
Signs and Symptoms
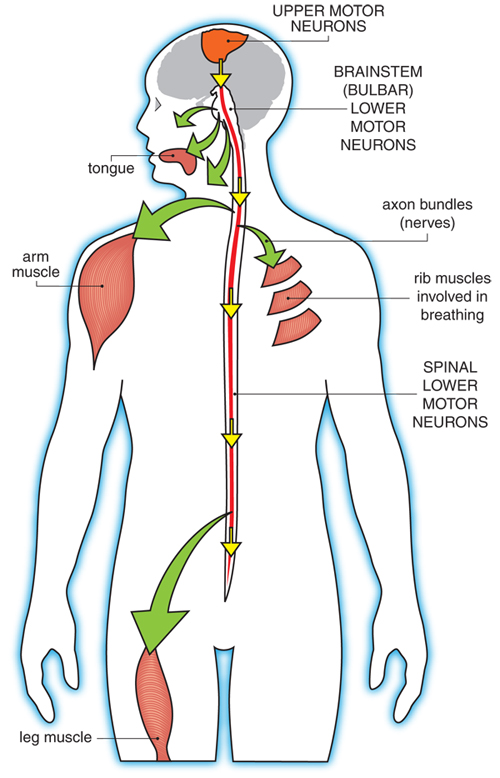
ALS affects the upper motor neurons, which are in the brain, and the lower motor neurons, which are in the spinal cord and brainstem. Upper motor neuron degeneration generally causes spasticity (tightness in a muscle), slowness of movement, and increased reflexes, while lower motor neuron degeneration causes muscle weakness, muscle atrophy (shrinkage of muscles) twitching (fasciculations), cramps, and reduction or loss of reflexes. These can occur in combination in ALS, as upper and lower motor neurons are affected at the same time. It is important to note that every voluntary muscle has both an upper and a lower motor neuron.
ALS can affect people of any age, though it usually strikes in late middle age. ALS typically announces itself with persistent weakness or spasticity in an arm or leg (80% of all cases), causing difficulty using the affected limb. Sometimes (in about 20% of all cases) the problem presents first in the muscles controlling speech, producing alterations in the vocal quality, or swallowing, which may lead to coughing and choking. ALS can even manifest as inappropriate laughing, crying, or yawning (pseudobulbar affect). Respiratory onset with shortness of breath as the presenting symptom is the rarest presentation.
It is not unusual for people to ignore such problems for some time at this stage, or to consult a physician who may be relatively unconcerned. However, the disease, if it is truly ALS, generally spreads from one part of the body to another so that eventually the problem can no longer be ignored. It is at this point that people usually are referred to a neurologist, who will consider ALS among many other possible diagnoses.
Cardiac muscle and smooth muscle, which controls the digestive system, urinary tract and sexual function are not directly affected in ALS. Constipation may occur as a side effect of immobility and altered food and water intake but is not due to a direct failure of the GI tract. Hearing, vision, smell, taste, and touch generally remain normal. Some patients complain about excessive sweating, but the association between sweating and ALS remains unclear.
Pain can occur as a result of immobility and its various complications, especially if precautions such as daily range-of-motion exercises are not undertaken. Also pain due to nerve affection may occur in some patients with ALS.
Fasciculations are a common symptom of ALS. These persistent muscle twitches are generally not painful but can interfere with sleep. They are the result of the ongoing disruption of signals from the nerves to the muscles that occurs in ALS.
Some with ALS experience painful muscle cramps, which can sometimes be alleviated with medication.
Some people with ALS undergo alterations in their thinking or may exhibit uncharacteristic behavior changes, often referred to as frontotemporal dementia, or FTD. However, memory loss, a hallmark of Alzheimer's-type dementia, is generally not a feature of ALS. Instead, the person with ALS might be irritable, inconsiderate, apathetic, or impulsive or might otherwise act in uncharacteristic ways. An alternative presentation of FTD is a loss of language skills that in the extreme can turn into a loss of all language meaning.
Another potential ALS symptom — not experienced by all — is a temporary lapse of control over emotional expressions such as laughing or crying, a phenomenon called pseudobulbar affect. Laughing or crying bouts, often triggered by the smallest of things, are more related to the disease process rather than to actual feelings of happiness or sadness. There are medications, including Nuedexta, that can be effective in treating this symptom, if it is bothersome to the patient.
Additional reading
- Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, Sobue G. Amyotrophic lateral sclerosis. Lancet. 2022 Oct 15;400(10360):1363-1380. doi: 10.1016/S0140-6736(22)01272-7. Epub 2022 Sep 15. PMID: 36116464; PMCID: PMC10089700.
- Siddique N, Siddique T. Amyotrophic Lateral Sclerosis Overview. 2001 Mar 23 [Updated 2023 Sep 28]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1450/
Last revised August 2025
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Grants at a Glance
Read More -

Search for Clinical Trials
Learn More

