Amyotrophic Lateral Sclerosis (ALS)
Medical Management
Medical interventions and technology have vastly improved the quality of life for people with ALS by assisting with breathing, nutrition, mobility, and communication. Proper management of symptoms and proactive use of medical interventions and equipment can make a positive difference in day-to-day living and potentially may lengthen life.
People with ALS should discuss any medical or mental health concerns with their ALS doctor. Living well with ALS means doing everything possible to cope with symptoms as they occur, if not before.
Managing the symptoms is the mainstay treatment for ALS; multidisciplinary care may improve quality of life.
ALS Care Guidelines
The American Academy of Neurology (AAN) released revised physician guidelines for medical management of ALS in October 2009. The guidelines include information about drug, nutritional, and respiratory therapies; multidisciplinary care; symptom management; and cognitive/behavioral impairment. Since the release of the AAN guidelines, several therapies for ALS have been approved by the US Food and Drug Administration (FDA) that may help manage symptoms, reduce the rate of decline, or prolong survival.
Medications
The drug riluzole (Rilutek) was approved by the FDA to treat ALS in December 1995. It is an oral medication that may reduce motor neuron damage by decreasing levels of glutamate (molecule involved in signaling between nerve cells and motor neurons). Rilutek has a modest effect on slowing disease progression and prolonging life. In clinical trials, Rilutek prolonged survival in people with ALS by three to six months. The drug appears to be safe, but it is expensive, and can cause fatigue, nausea, and liver damage. The manufacturer suggests that people taking Rilutek should avoid excessive intake of alcohol to minimize the risk of liver damage.
Newer formulations of riluzole allow the drug to be easier to take for individuals who have difficulty swallowing. Tiglutik is a thickened liquid formulation of riluzole that was approved by the FDA in September 2018. Exservan is dissolvable tablet formulation of riluzole that was approved in November 2019.
In October 2010, dextromethorphan HBr and quinidine sulfate (Nuedexta) was approved specifically for an aspect of ALS called pseudobulbar affect (PBA) in which affected people experience involuntary outbursts of laughing or crying. Nuedexta has been shown to reduce the rate of daily episodes in people experiencing PBA.
Edaravone (Radicava) is an antioxidant therapy that received FDA approval in May 2017. Radicava is given intravenously (IV) and is thought to protect neurons from the damage caused by oxidative stress (toxicity due to harmful free radicals). Edaravone was shown in clinical trials to slow functional decline in some people with ALS. RADICAVA ORS is a form of edaravone taken orally or by feeding tube rather than by IV. RADICAVA received FDA approval in May 2022.
Most recently, on April 25, 2023, the FDA granted accelerated approval of tofersen (Qalsody) for the treatment of ALS associated with mutation in the superoxide dismutase 1 (SOD1) gene (SOD1-ALS). Qalsody is the fourth approved therapy to treat a form of ALS and the first therapy to target a genetic cause of ALS. Qalsody is a type of drug known as an antisense oligonucleotide (ASO) and it acts by reducing the production of new SOD1 protein in cells. In clinical trials, treatment with Qalsody was shown to lower levels of plasma neurofilament light (NfL), a biomarker of nerve injury and neurodegeneration, as well as SOD1 protein levels, in treated participants.
In addition to these disease-modifying treatments, physicians may prescribe medications to treat symptoms of ALS including muscle cramps and stiffness, drooling, anxiety and depression, constipation (the result of reduced mobility and/or weakened abdominal muscles), sleep difficulties, and pain associated with prolonged immobility.
Multidisciplinary care
The AAN guidelines found that visiting a multidisciplinary ALS clinic (one with many types of health professionals, such as an MDA/ALS Care Center) can help people with ALS get the best possible care. The evidence showed that people with ALS who get care at a multidisciplinary clinic live longer and may have a better quality of life than those who do not.
An ALS care team might include physicians, pharmacists, physical, occupational, speech, and respiratory therapists, clinical psychologists, nutritionists, social workers, and home care and hospice nurses, among other professionals. This team can help create a personalized treatment plan and recommend medications and adaptive equipment to keep people with ALS comfortable, mobile, and independent for as long as possible.
Breathing
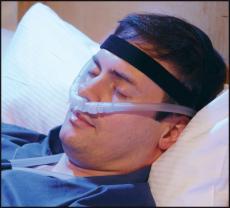
The diaphragm is an arched muscle located just beneath the lungs that moves up and down and allows air to come in and move out. The intercostals are muscles between the ribs that contract and relax and also assist with air movement. As the diaphragm and intercostal muscles weaken in ALS, the act of breathing, which is entirely automatic for most people, becomes conscious and energy-consuming.
To preserve quality of life (and to prolong life itself in later stages of ALS) introducing preventive measures to maintain breathing becomes necessary. Such symptoms as the inability to cough, shortening of spoken sentences, daytime headaches, sleepiness, exhaustion, and weight loss may indicate that breathing problems have advanced to the point where respiratory support will be valuable.
A pulmonologist and respiratory therapist typically are involved in managing the care of individuals with ALS. Shortly after an ALS diagnosis is made, many specialists recommend a breathing test called a forced vital capacity (FVC) and other pulmonary function tests (PFTs). In the event that a patient presents with weakness in the muscles of their mouth, another breathing test called maximal sniff nasal inspiratory force (SNIF, also called the sniff nasal pressure), avoids the need for a mouthpiece. These tests give the clinic team baseline measures against which later breathing tests can be compared.
The physician may recommend noninvasive ventilation to compensate for weakened muscles by assisting the movement of air in and out of the lungs. Generally, supplemental oxygen is not prescribed for individuals with ALS unless there are other medical conditions that require it.
Oxygen may even do harm, so it should be used with caution, if at all. Please inform your physician of your condition when admitted to a hospital.
Noninvasive ventilation comes in many forms but usually consists of two basic elements: an “interface,” such as a mask or nose inserts, and air delivered under pressure by a small, portable machine. Usually, there is one pressure for inhalation and another pressure for exhalation. This type of machine is often called a BiPAP (a registered trademark of Philips Respironics) for bilevel positive airway pressure.
The device does not necessarily require around-the-clock use. Pressures, masks, and other aspects of the device can be changed by the medical team as needed. Ideally, the person with ALS should try several interface options (full-face masks, nasal pillows, etc.) and practice breathing through them to see which are the most comfortable.
The most permanent type of ventilation is the positive-pressure ventilator with a surgically created tracheostomy or trach. A ventilator is attached by a breathing hose to a tracheostomy tube that delivers air through the neck into the trachea (windpipe) on a timed cycle.
Tracheostomy surgery (to create an opening in the trachea) is usually followed by several days or weeks of rehabilitation, during which caregivers learn how to clean and maintain the tracheostomy tube, change supplies, and perform suctioning of mucus.
Many people on total ventilatory support can continue working, traveling, socializing, and enjoying life. Today’s vents are small, portable, relatively quiet, and can be carried on a wheelchair. For those still able to speak, a speaking valve often can be added to the inflatable cuff at the end of the tracheostomy tube, allowing air to travel to the vocal cords and enable speech.
In addition, a tracheostomy can provide a great feeling of safety. Permanent vents have alarms to alert caregivers to congestion or a disconnected tube. And invasive ventilation can return some energy as it relieves exhaustion caused by poor sleep, prolonged coughing, and labored breathing.
Another aspect of respiratory care that’s important in ALS is assisted coughing. As the coughing muscles weaken, it becomes harder to clear mucus from the airways and life-threatening mucus plugs can form. An assisted coughing device, which pushes air into the airways through a mask and then quickly reverses air flow, can help clear the airways and prevent infection. Doctors may recommend other methods to assist with coughing and clearing secretions from the airways.
It is recommended that patients with ALS receive pneumococcal vaccination and annual seasonal influenza vaccination as they may have compromised ability to handle respiratory secretions and an increased risk of developing chronic pulmonary conditions.
Cognitive and behavior changes
Although ALS is considered a disease of the motor (movement) system, cognitive (thinking) and behavioral changes can also occur in this disease. Some changes may be in response to the devastating nature of the disease, while others appear to be neurological in origin and part of the disease process itself.
Many patients do experience alterations in thinking or behavior, although in most instances, symptoms are minimal and may be more distressing to relatives and caregivers than to the affected person. Although the medical profession often refers to these changes as frontotemporal dementia (FTD), which connotes memory loss (as in Alzheimer's disease), memory is generally well-preserved in ALS. Instead, the person with ALS may become unduly angry or irritable or may be less considerate of others than one might expect them to be, or may exhibit poor judgment, apathy, ritualistic habits, new dietary preferences, or other uncharacteristic behavior.
ALS is related to frontotemporal dementia (FTD). Both are progressive neurodegenerative diseases, characterized by degeneration of the frontal and temporal lobes of the brain. ALS and FTD lead to a disturbance in behavior, personality, and language. It is estimated that 50% of ALS patients show signs of behavioral dysfunction or subtle cognitive impairment, like dementia. About 15% of patients diagnosed with ALS reach the diagnostic criteria of FTD and are referred to as having ALS-FTD.
Some people with FTD symptoms lose insight into their actions and may not realize (even when it is pointed out to them) that they are not thinking as clearly as before or that their behavior may change.
It is important for caregivers to know about possible cognitive symptoms in ALS so that they can recognize the signs and not think that their loved one is simply "being difficult." Families can deal with the cognitive and behavioral changes associated with FTD by making modifications to the environment of the person with ALS that improve safety and organization; carefully describing symptoms to healthcare providers; helping their loved one make important decisions as early in the disease process as possible; and seeking support from others affected by ALS through online or in-person groups.
Pseudobulbar affect
Another phenomenon that sometimes occurs in ALS is known as pseudobulbar affect (PBA). A patient who experiences PBA presents with sudden uncontrollable outbursts of laughing or tearing, without the ability to control them. Half of patients present with PBA symptoms, mainly those who are diagnosed with the bulbar form of ALS. In 2010, the drug combination dextromethorphan HBr and quinidine sulfate (Nuedexta) was approved to treat this symptom. Other drugs used to treat this condition include antidepressants.
Communication

Speaking ability is lost when ALS affects the muscles of the mouth and throat that control speech and the muscles that help move air over the vocal cords.
For this reason, speech therapists and speech-language pathologists are vital members of the ALS care team. A speech therapist can teach the person with ALS special techniques for conserving energy and making speech more understandable. In some cases, a dentist can make a device called a palatal lift that can help compensate for certain types of weakness in the roof of the mouth.
In the early stages of ALS, speech therapists may suggest voice banking, which involves recording a number of common phrases that later can be programmed into a computer or communication device, enabling individuals with ALS to continue speaking in their own voice when they communicate via assistive technology.
Electronic assistive communication devices controlled by hand or eye movement are also an option.
Many therapists recommend being proactive about exploring communication options well before assistive technology is needed. A speech therapist can demonstrate how to use an alternative augmentative communication device, or AAC device.
Because learning to use such a device at later stages of ALS may be harder, patients are encouraged to discuss options with their care team and speech therapist early on, in order to proactively plan ways to stay in the conversation.
Cramps, twitches, and spasticity
Muscle cramps, twitches, and spasticity (tightness) are common in ALS.
Mexiletine is a drug used to reduce frequent and painful cramps. Other drugs used to treat muscles spasms are levetiracetam, carbamazepine, and phenytoin.
Other effective treatments for these problems include baclofen, dantrolene, and tizanidine. These medications require a doctor's prescription, after considering possible side effects.
Rest, repositioning, heat (such as from a microwaveable pad or warm bath), or gentle massage often are helpful in relieving the discomfort associated with muscle cramps, twitching, and tightness.
Pain
The damage ALS causes to the nervous system can lead to uncomfortable symptoms such as cramps and muscle twitching, which can be painful. Also, the immobility that results from ALS may put stress on muscles and joints, causing what is known as secondary pain. Reduced mobility can cause pressure-induced skin injury.
Management options for pain include the use of anti-inflammatory drugs and opioids.
Depression
There have been no controlled trials of treatment for depression in ALS, although there is consensus among experts that it should be treated. Many clinicians have found that antidepressants or anti-anxiety medication can have a positive effect.Studies have estimated the prevalence of depression in people with ALS to be approximately 33% or less. According to the 2009 ALS Care Guidelines, there have been no controlled trials of treatment for depression in ALS, although there is consensus among experts that it should be treated. Many clinicians have found that antidepressants or anti-anxiety medication can have a positive effect.
Drooling
Drooling, also known as sialorrhea, is common in ALS because of weakness of the mouth and throat muscles. The October 2009 AAN ALS Care Guidelines say that the oral medication amitriptyline may be helpful in drying up saliva (thereby reducing drooling), as may injections of botulinum toxin type B into the salivary glands. Other drugs that can be used to treat drooling are atropine, hyoscyamine, and glycopyrrolate.
Radiation of the salivary glands to reduce saliva production has also been used for treatment of this condition when other measures fail. However, the guidelines note that side effects may include redness, sore throat, and nausea.
Eating, drinking, and nutrition
Evidence shows that maintaining one's weight may increase survival with ALS. Severe weight loss means muscle loss. Additionally, adequate fluid intake is essential for hydration, keeping saliva and mucus thin, and avoiding constipation.
Swallowing difficulties (dysphagia) are a prime cause of weight loss. As the muscles involved in chewing, moving food toward the back of the mouth, and swallowing weaken in ALS, eating and drinking become less pleasurable and more hazardous and time-consuming. Mouth and throat weakness can lead to choking and aspiration (inhaling food or liquid into the lungs), which can cause respiratory infection. Changing the consistency of food is the initial management for swallowing difficulties.
Other challenges include arm/hand weakness that limits self-feeding, decreased appetite, constipation, shortness of breath and nausea after eating, or fatigue due to the long and tiring process of eating.
Speech-language pathologists or therapists are also specialists in swallowing, as these functions involve the same muscles as speech. Some therapists specialize more in speech and others more in swallowing. Early solutions involve changing the consistency of food and liquids — usually thickening the liquids and avoiding large pieces of food — as well as changing swallowing techniques.
If swallowing becomes difficult or unsafe and/or if eating takes a great deal of time and energy, a feeding tube may be recommended, often called a gastrostomy tube, g tube or PEG (percutaneous endoscopic gastrostomy) tube. The term "gastrostomy" refers to making a small incision in the stomach. It is usually done percutaneously, which means “through the skin,” with the help of an endoscope, a medical instrument.
The October 2009 AAN report found no ALS-specific indications for the timing of feeding tube placement. However, the report said the risk of placement increased when respiratory function declined below 50% of normal and suggested that those with swallowing difficulties will be exposed to less risk if a feeding tube is placed when respiratory function is above 50% of normal.
If it is still possible to swallow some foods or liquids safely, the person with ALS can continue to eat and drink by mouth even after placement of a feeding tube. But eating by mouth is no longer the only way to get adequate nutrition. This can be a relief to those who cannot take in enough calories by mouth because they get too tired or are afraid of choking or aspirating food.
Although getting a feeding tube may initially feel like "ALS is winning," those who get them find they regain time and energy and reduce the strain on their caregivers.
Supplementation
Vitamin supplements may be recommended if swallowing difficulties result in reduced intake of nutrients. Reasonable doses of antioxidant supplements, such as vitamins C and E, are thought to have beneficial effects on the nervous system and overall health. However, high doses can do more harm than good. A person’s physician can help determine which medications and/or vitamin supplements may be beneficial and discuss dosage levels of supplements.
Hand function
Occupational therapists specialize in helping people find and use tools to cope with progressive weakness in hand muscles. Special grips for writing and eating utensils, devices that fit over keys to make them easier to turn, zipper pulls, and button hooks can help make weakening hands more functional and help preserve independence in activities of daily living. Consult with the occupational therapist on your MDA Care Center team for more information.
Mobility
Fatigue, falling, and increased difficulty walking often are experienced as ALS progresses. Avoiding falls is of paramount importance and can prevent trauma that could accelerate ALS disease progression. In addition to using mobility equipment to avoid falls, be sure to move area rugs, install grab bars, and eliminate clutter wherever possible. Carry a cell phone when walking alone to call for help if necessary. Reduced mobility can cause musculoskeletal pain as well as pressure-induced skin and soft tissue injury.
In the early stages of ALS, mobility equipment such as a cane, walker, or supportive brace (orthosis) can provide help in getting around. Weakness of muscles controlling the foot make it hard to move the foot or toe at will, causing foot drop that can lead to trips and falls. A lightweight ankle-foot orthosis (AFO) keeps the foot from dropping and adds steadiness when walking. It can be slipped into different styles of shoes and concealed by wearing a sock over the brace, but it’s best worn with a supportive tie-up shoe.
When walking becomes difficult, riding in a manual wheelchair for long distances can conserve energy for short distance walking and also help prevent injury.
In later stages of the disease, a power wheelchair is usually the preferred means of mobility. When the time comes to transition to a power wheelchair full time, many with ALS find that they recover a large degree of independence.
Power chairs can be driven by a variety of means besides by hand, including by eye gaze and by "sip and puff" breath control. A “tilt-in-space” option on a wheelchair allows the seat to be positioned at a variety of angles, relieving pressure, and helping prevent skin breakdown. Other valuable power chair options include elevating seats, chairs that turn into standers, motorized leg rests, custom seating, and more.
Physical therapists, occupational therapists, and equipment specialists have specialized knowledge about maintaining mobility and using equipment and should be consulted prior to buying. Remember that insurance often will pay for only one mobility device during a set period of time, so it is important to consider upcoming needs before buying. If possible, it may be wise to borrow interim equipment, such as a mobility scooter or manual wheelchair, and save insurance for big-ticket items like a power wheelchair.
Weakness in neck muscles is also associated with ALS. This results in difficulties in controlling or holding up the head, which leads to excessive fatigue and discomfort, not to mention frustration. Cervical collars may be recommended for support.
Very little research has been done on the subject of exercise and its role in ALS. In fact, it is not known whether exercises are beneficial for increasing muscle strength for people with ALS.
However, it is widely accepted among physicians and therapists that specific kinds of exercise help prevent the development of painful contractures (the permanent tightening of muscles) and can decrease the spasticity (intermittent or constant muscle tightness or spasms) common in ALS.
Practicing the healthiest type of exercise for each stage of ALS will help maintain comfort and mobility. For some people, a moderate amount of daily walking in the early stages of ALS may be advisable. As the disease advances, it's very important to do daily range-of-motion and stretching exercises, either independently or with the help of a caregiver. A physical therapist can help determine the best exercise regime.
Planning early for the inevitable changes that occur over the course of ALS goes a long way toward maintaining function and independence for as long as possible and maintaining the highest possible quality of life. Such plans include addressing how home accessibility may be affected as the disease progresses. There are many ways to make adaptations or modifications that promote independence and safety at home. A home visit by an occupational therapist to assess the environment is very helpful.
Additional reading
- Miller RG, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009 Oct 13;73(15):1227-33. doi: 10.1212/WNL.0b013e3181bc01a4. PMID: 19822873; PMCID: PMC2764728.
- Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, Sobue G. Amyotrophic lateral sclerosis. Lancet. 2022 Oct 15;400(10360):1363-1380. doi: 10.1016/S0140-6736(22)01272-7. Epub 2022 Sep 15. PMID: 36116464; PMCID: PMC10089700.
- Siddique N, Siddique T. Amyotrophic Lateral Sclerosis Overview. 2001 Mar 23 [Updated 2023 Sep 28]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1450/
Last revised August 2025
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Grants at a Glance
Read More -

Search for Clinical Trials
Learn More

