Mitochondrial Myopathies (MM)
Causes/Inheritance
What causes mitochondrial diseases?
Genetic origins
Mitochondrial myopathies are among the more common inherited metabolic muscle disorders, occurring in about 1 in 5,000 individuals.
Mitochondrial diseases are not contagious and not caused by lifestyle or behavior. They result from genetic mutations that disrupt the production of proteins essential for normal mitochondrial function. These mutations are inherited from parents to children, making mitochondrial diseases heritable, though the specific symptoms and severity can vary widely among family members.
How mitochondria normally produce energy through oxidative phosphorylation
Genes involved in mitochondrial function encode proteins that form a highly efficient energy-producing system inside each mitochondrion. This system converts sugars and fats into adenosine triphosphate (ATP) through oxidative phosphorylation, which relies on oxygen.
Early in the process, specialized proteins import sugars and fats into the mitochondrion. Other proteins then break down these molecules and extract electrons, which move through a sequence of protein complexes known collectively as the electron transport chain. These complexes—labeled I through IV—work alongside coenzyme Q10 and cytochrome c to shuttle electrons. The energy generated by this movement is then used by complex V, also called ATP synthase, to produce ATP.
How mutations cause mitochondrial disease
Deficiencies in key proteins: A deficiency in one or more components of the oxidative phosphorylation pathway can cause mitochondrial disease. For example, complex I deficiency is a well-recognized cause. Some conditions, such as coenzyme Q10 deficiency due to nuclear DNA mutations, can also present as isolated muscle weakness.
Problems with protein processing and mitochondrial structure: For oxidative phosphorylation to work correctly, proteins must be accurately produced, transported into the mitochondria, and inserted into the inner mitochondrial membrane. Mutations that interfere with any of these steps can impair energy production. Mutations can also affect the structure and dynamics of the mitochondria themselves. When many defective mitochondria accumulate, the cell becomes deprived of ATP and begins to accumulate unused fuel molecules and reactive oxygen.
Consequences of mitochondrial dysfunction
Toxic byproducts and cellular stress: When normal energy production breaks down, cells switch to less efficient methods of generating ATP. This shift leads to lactic acid buildup, known as lactic acidosis, which contributes to muscle fatigue and can damage muscle and nerve tissue. Meanwhile, excess oxygen can be converted into reactive oxygen species, including free radicals, which further harm cells.
Effects on muscle and nerve cells: ATP derived from mitochondria provides the main source of power for muscle cell contraction and nerve cell firing. So, muscle cells and nerve cells are especially sensitive to mitochondrial defects. The combined effects of energy deprivation and accumulation of toxic byproducts in these cells give rise to the main symptoms of mitochondrial myopathies and encephalomyopathies.
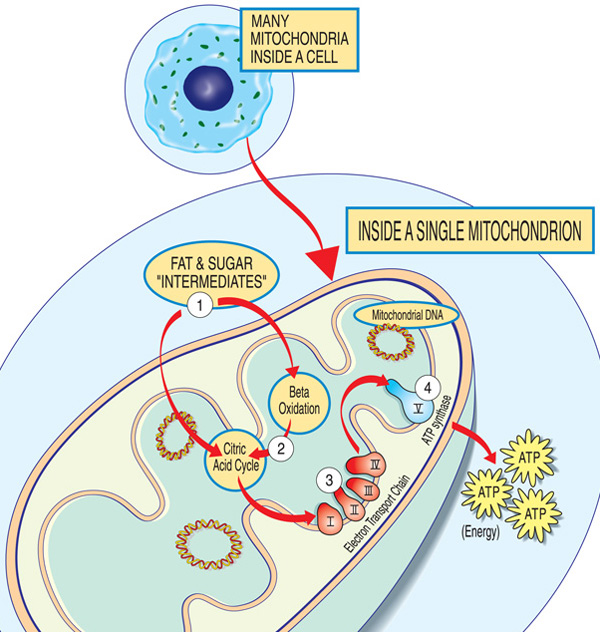
Each mitochondrion is an energy factory that “imports” sugars and fats, breaks them down and “exports” energy (ATP) via these steps: Fat and sugar intermediates enter the mitochondrion. Fatty acids are broken down through beta oxidation and the removal of electrons in the citric acid cycle. Electrons are passed through the major complex of the electron transport chain. ATP is made by ATP synthase.
What are the inheritance patterns in mitochondrial myopathies?
Mitochondrial genetics are complex. While mitochondrial diseases are caused by defective genes and run in families, they can be difficult to trace through a family tree.
To understand the inheritance of mitochondrial diseases, it is important to know that there are two types of genes essential to mitochondria. The first is housed within the nucleus — the part of our cells that contains most of our genetic material, or DNA. The second resides exclusively within DNA contained inside the mitochondria themselves.
Mutations in either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA) can cause mitochondrial disease.
Most nDNA, along with any mutations it carries, is inherited in a Mendelian pattern, with one copy of each gene coming from each parent. Many mitochondrial diseases caused by nDNA mutations, including MNGIE and MDS, follow an autosomal recessive pattern, in which mutations in both copies of a gene are required to cause disease.
Unlike nDNA, mtDNA passes only from mother to child. That is because during conception, when the sperm fuses with the egg, the sperm’s mitochondria — and its mtDNA — are destroyed. Thus, mitochondrial diseases caused by mtDNA mutations are inherited in a maternal pattern (see illustration below).
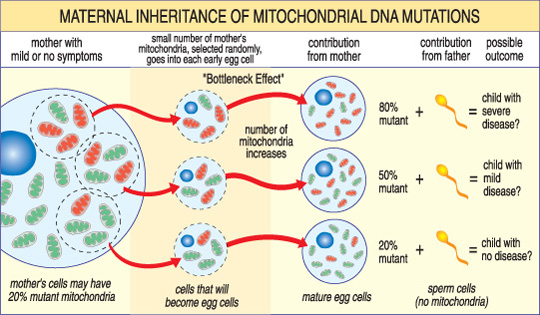
The severity of a mitochondrial disease in a child depends on the percentage of abnormal (mutant) mitochondria in the egg cell that formed them.
A unique feature of mtDNA diseases arises from the fact that a typical human cell — including the egg cell — contains only one nucleus but hundreds of mitochondria. A single cell can contain both mutant mitochondria and normal mitochondria, and the balance between the two will determine the cell’s health. This helps explain why the symptoms of mitochondrial disease can vary so much from person to person, even within the same family.
For example, a woman’s egg cells may have different proportions of mutant mitochondria; a child from an egg with mostly healthy mitochondria may be unaffected, while a child from an egg with mostly mutant mitochondria is likely to develop disease. The mother’s own symptoms may also vary depending on her mitochondrial mix.
The risk of passing on a mitochondrial disease to children depends on many factors, including whether the disease is caused by mutations in nDNA or mtDNA. A good way to find out more about these risks is to talk to a doctor or genetic counselor.
In addition to being inherited, mitochondrial diseases can also arise in a sporadic fashion, meaning they may occur with no family history.
To learn more, see Print-Ready Educational Materials.
Additional reading
- Ahuja AS. Understanding mitochondrial myopathies: a review. PeerJ. 2018 May 21;6:e4790. doi: 10.7717/peerj.4790.
- Gorman, GS et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015. doi:10.1002/ana.24362
- Wen H et al. Mitochondrial diseases: from molecular mechanisms to therapeutic advances. Signal Transduct Target Ther. 2025 Jan 10;10(1):9. doi: 10.1038/s41392-024-02044-3.
Last reviewed May 2026.
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Search for Clinical Trials
Learn More -

Grants at a Glance
Read More

