About Us
Our mission is to empower people living with neuromuscular diseases to live longer, more independent lives.
Sarepta Therapeutics Reports Long-Term Outcomes through 144 Weeks from Phase IIb Study of Eteplirsen in Duchenne Muscular Dystrophy
CAMBRIDGE, Mass. – July 10, 2014 – Sarepta Therapeutics, Inc. (NASDAQ: SRPT), a developer of innovative RNA-based therapeutics, today announced data through Week 144 from Study 202, a Phase IIb open-label extension study of eteplirsen in patients with Duchenne muscular dystrophy (DMD). After nearly three years of follow up, results on the 6-minute walk test (6MWT) showed a decline in walking ability at a rate slower than would be expected based on available DMD natural history data. In addition, a continued stabilization of respiratory muscle function was observed, as assessed by pulmonary function tests. As previously reported, Study 202 met its primary endpoint of increased novel dystrophin as assessed by muscle biopsy at Week 48 and is now in the long-term extension phase in which patients continue to be followed for safety and clinical outcomes.
At Week 144, patients in the 30 mg/kg and 50 mg/kg eteplirsen cohorts who were able to perform the 6MWT (modified Intent-to-Treat or mITT population; n=6) experienced a decline of 33.2 meters, or about 8.5 percent, from baseline in walking ability. A statistically significant treatment benefit of 75.1 meters (p≤0.004) was observed for the mITT population compared with the placebo/delayed-treatment cohort (n=4), which initiated treatment at Week 25 following 24 weeks of placebo. After experiencing a substantial decline of 68.4 meters from baseline to Week 36, the placebo/delayed-treatment cohort demonstrated a decline of 39.0 meters in walking ability from Week 36 through Week 144, the period from which meaningful levels of dystrophin were likely produced. These analyses were based on the maximum 6MWT score when the test was performed on two consecutive days.
“The long-term clinical data for eteplirsen showing a slowing in the decline of walking ability in a population now on average 12 years old are very encouraging, particularly when compared with the growing body of DMD natural history data which clearly show that similarly aged patients typically experience an increasingly rapid decline in walking ability and lose ambulation in their early teen years,” said Jerry Mendell, M.D., director of the Centers for Gene Therapy and Muscular Dystrophy at Nationwide Children's Hospital and principal investigator of the Phase IIb study.
“We now have nearly three years of treatment experience with eteplirsen from our Phase IIb clinical study program and, based on guidance from the U.S. Food and Drug Administration earlier this year, we plan to submit these results along with additional data and analysis as part of a New Drug Application for eteplirsen by year-end,” said Chris Garabedian, president and chief executive officer of Sarepta Therapeutics.
Respiratory muscle function from baseline through Week 144 in the Intent-to-Treat population (n=12), as measured by maximum inspiratory and expiratory pressure (MIP and MEP), showed a 14.7 percent mean increase in MIP and a 12.8 percent mean increase in MEP. Analyses of MIP percent predicted (MIP adjusted for weight) and MEP percent predicted (MEP adjusted for age) demonstrated a mean change from 91.7 percent at baseline to 93.9 percent at Week 144 in MIP percent predicted, and a mean change from 79.3 percent at baseline to 75.7 percent at Week 144 in MEP percent predicted. In addition, there was a mean increase in forced vital capacity (FVC), a measure of lung volume, of 11.0 percent. FVC percent predicted (FVC adjusted for age and height) was maintained above a mean of 90 percent at Week 144, with 101.3 percent at baseline and 90.9 percent at Week 144.
“We are encouraged to see continued stability on measures of respiratory muscle function in patients treated with eteplirsen for nearly three years, particularly as declines in MIP and MEP are often the first signs of pulmonary dysfunction in DMD,” said Edward Kaye, M.D., senior vice president and chief medical officer of Sarepta Therapeutics. “As we prepare to submit a New Drug Application for eteplirsen including these data, we are also on track to initiate in the coming months several new clinical studies of eteplirsen in a broader patient population to further characterize the drug’s safety and efficacy profile.”
Through 144 weeks, eteplirsen was well tolerated and there were no reported clinically significant treatment-related adverse events and no treatment-related serious adverse events. In addition, there were no treatment-related hospitalizations or discontinuations.
Summary of 6MWT: Week 144 Treatment Results*
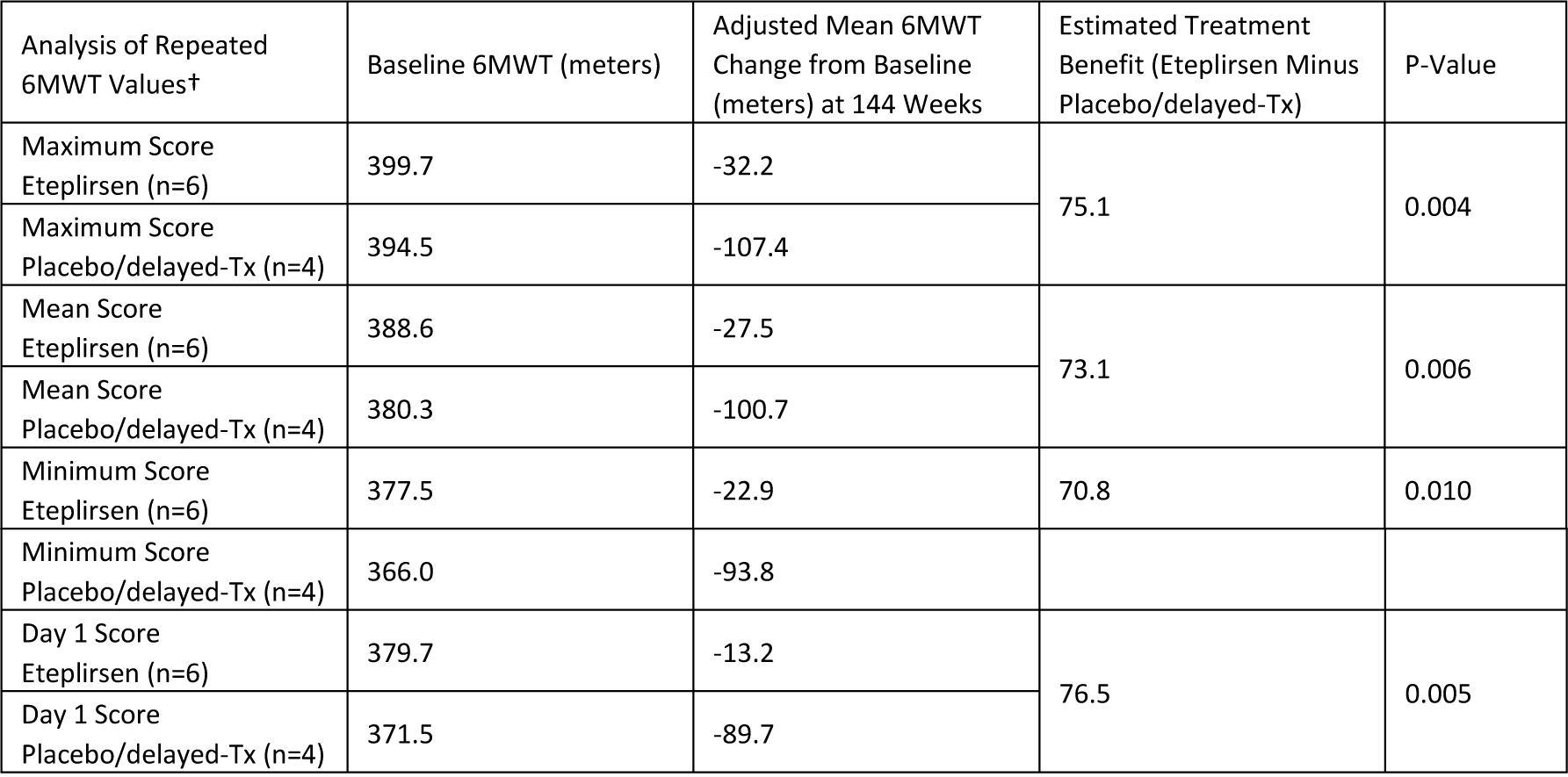
* All 6MWT analyses are based on a Mixed Model Repeated Measures test.
† All 6MWT analyses include the mITT population
‡ The pre-specified primary analysis of the 6MWT results was based on the maximum score.
Patients performed two 6MWT evaluations on consecutive days at time points coinciding with a muscle biopsy procedure at baseline and Weeks 12, 24 and 48. Two 6MWT evaluations were also performed at Weeks 120 and 144, and will be performed at all future functional assessment visits. All other evaluations were a single 6MWT. The pre-specified primary analysis included the maximum distance walked at those clinic visits where repeated tests were taken. Other analyses of the repeated 6MWT results assessed mean, minimum, and Day 1 (first measure) scores. Results from these additional 6MWT analyses confirm the data observations in the primary analysis.
Summary of Pulmonary Function Tests: Week 144 Treatment Results
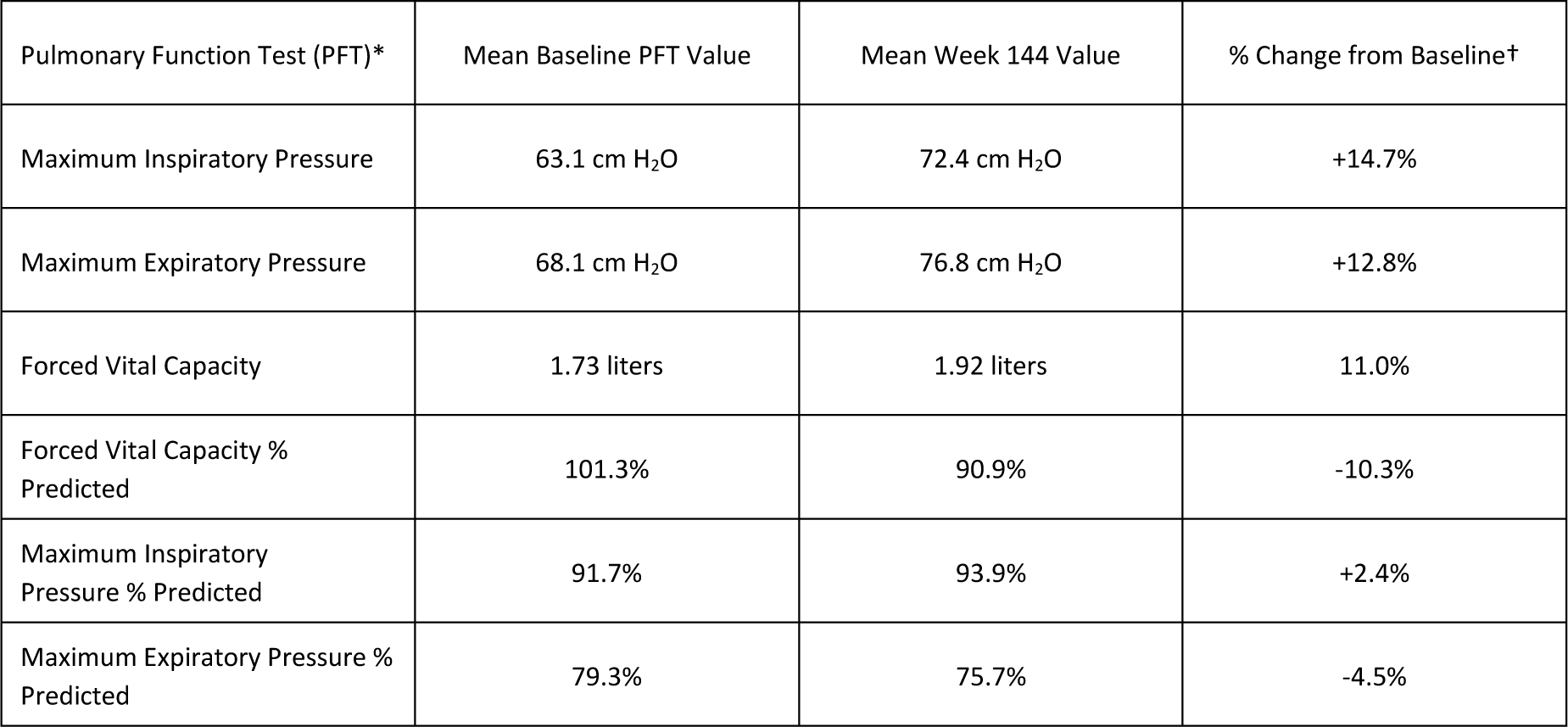
* All PFT analyses include the ITT population (N=12)
† All Week 144 data were not statistically significantly different from baseline, except for a statistically significant increase & decrease in FVC & FVC % Predicted, respectively (using one-sample t-test).
Summary of Additional Exploratory Efficacy Endpoints
Results through Week 144 for other exploratory efficacy endpoints, including timed function tests (e.g., Gowers’ maneuver, 10 meter run/walk and timed 4-step test) and the North Star Ambulatory Assessment have shown continued declines compared to baseline, though at potentially slower rates as compared to the limited available natural history data. These endpoints are less well characterized in DMD patients than the 6MWT and pulmonary function tests and have more inter- and intra-patient variability, although they may be predictors of decline at various stages of this disease. All patients evaluable on measures of ambulation (modified Intent-to-Treat, or mITT population) are still able to perform these tests including the 10 meter run/walk and 4-step test, with the exception of two patients who are no longer able to perform the Gowers’ maneuver.
About the Phase IIb Eteplirsen Program (Studies 201 and 202)
Study 201 was a randomized, double-blind, placebo-controlled clinical study conducted at Nationwide Children’s Hospital in Columbus, Ohio. Twelve boys aged 7 to 13 years with a confirmed genotype amenable to treatment with an exon-51 skipping drug were randomized to one of three cohorts: 30 mg/kg (n=4), 50 mg/kg (n=4), and placebo/delayed treatment (n=4). Eteplirsen and placebo were administered weekly by intravenous infusion.
At Week 25, all patients rolled over to Study 202, a long-term open-label extension study, and placebo-treated patients initiated eteplirsen treatment at 30 mg/kg (n=2) or 50 mg/kg (n=2).
The primary efficacy endpoint in Study 201 and Study 202 was the increase in novel dystrophin as assessed by muscle biopsy at Weeks 12 and 24 and at Week 48, respectively. The primary clinical endpoint was the 6MWT, a well-accepted measure of ambulation and clinical function in DMD. Long-term follow up in Study 202 continues to evaluate safety and clinical outcomes including the 6MWT.
About the 6-Minute Walk Test (6MWT)
The 6-minute walk test (6MWT) was developed as an integrated assessment of cardiac, respiratory, circulatory, and muscular capacity for use in clinical trials of various cardiac and pulmonary conditions.1 In recent years, the 6MWT has been adapted to evaluate functional capacity in neuromuscular diseases and has served as the basis for regulatory approval of a number of drugs for rare diseases, with mean changes in the 6MWT ranging from 28 to 44 meters.2,3,4 Additionally, published data from longitudinal natural history studies assessing dystrophinopathy, a disease continuum comprised of DMD and Becker muscular dystrophy, support the utility of the 6MWT as a clinically meaningful endpoint in DMD.5 These data show that boys with DMD experience a significant decline in walking ability compared to healthy boys over one year, suggesting that slowing the loss of walking ability is a major treatment goal.
About the 6MWT Statistical Methodology and the Modified Intent-to-Treat (mITT) Population
The Mixed Model Repeated Measures (MMRM) test was used for all statistical analyses of the 6MWT results. Baseline 6MWT scores and duration since DMD diagnosis were included as covariates.
The mITT population used in the 6MWT analyses consisted of 10 of the 12 enrolled patients, including 4 patients in the 50 mg/kg cohort, 2 patients in the 30 mg/kg cohort and 4 patients in the placebo/delayed-treatment cohort. Two patients in the 30 mg/kg cohort showed rapid disease progression upon enrollment and lost ambulation by Week 24, and thus were excluded since they were no longer evaluable for the 6MWT. All other data were analyzed for all 12 patients.
About the Pulmonary Function Tests (PFTs)
Progressive respiratory muscle dysfunction in patients with DMD typically leads to ventilation assistance and respiratory failure, and may ultimately be a significant factor in patient mortality.6 Measurements of respiratory function are important for tracking the course of the disease, as well as the evaluation of potential therapeutic interventions. Maximum inspiratory pressure (MIP), maximum expiratory pressure (MEP) and forced vital capacity (FVC) were included in the Phase IIb clinical studies of eteplirsen as exploratory clinical outcome measures.
MIP and MEP measure the highest level of pressure a person can generate during inhalation and exhalation, respectively, and are the most sensitive measures of respiratory muscle strength.7 Specifically, MIP is a sensitive measure of diaphragm muscle weakness. In addition, DMD natural history studies have shown a decline in MEP before changes in other pulmonary function tests, including MIP and FVC, suggesting MEP is an early indicator of respiratory dysfunction.8 FVC measures the total volume of air expelled during forced exhalation after maximum inspiration. In DMD, FVC increases concomitantly with physical growth until the early teens. However, as growth slows or is stunted by disease progression, and muscle weakness progresses, the vital capacity enters a descending phase and declines at an average rate of about 8 to 8.5 percent per year after 10 to 12 years of age.8,9 MIP percent predicted (MIP adjusted for weight), MEP percent predicted (MEP adjusted for age) and FVC percent predicted (FVC adjusted for age and height) are supportive analyses.
About Duchenne Muscular Dystrophy
DMD is an X-linked rare degenerative neuromuscular disorder causing severe progressive muscle loss and premature death. One of the most common fatal genetic disorders, DMD affects approximately one in every 3,500 boys born worldwide. A devastating and incurable muscle-wasting disease, DMD is associated with specific errors in the gene that codes for dystrophin, a protein that plays a key structural role in muscle fiber function. Progressive muscle weakness in the lower limbs spreads to the arms, neck and other areas. Eventually, increasing difficulty in breathing due to respiratory muscle dysfunction requires ventilation support, and cardiac dysfunction can lead to heart failure. The condition is universally fatal, and death usually occurs before the age of 30.
About Sarepta’s Proprietary Exon-Skipping Platform Technology
Eteplirsen is Sarepta's lead drug candidate and is designed to address the underlying cause of DMD by enabling the production of a functional internally deleted dystrophin protein. Data from clinical studies of eteplirsen in DMD patients have demonstrated a broadly favorable safety and tolerability profile and restoration of dystrophin protein expression.
Eteplirsen uses Sarepta's novel phosphorodiamidate morpholino oligomer (PMO)-based chemistry and proprietary exon-skipping technology to skip exon 51 of the dystrophin gene enabling the repair of specific genetic mutations that affect approximately 13 percent of the total DMD population. By skipping exon 51, eteplirsen may restore the gene's ability to make a shorter, but still functional, form of dystrophin from messenger RNA, or mRNA. Promoting the synthesis of an internally deleted dystrophin protein is intended to stabilize or significantly slow the disease process and prolong and improve the quality of life for patients with DMD.
Sarepta is also developing other PMO-based exon-skipping drug candidates intended to treat additional patients with DMD.
About Sarepta Therapeutics
Sarepta Therapeutics is focused on developing first-in-class RNA-based therapeutics to improve and save the lives of people affected by serious and life-threatening rare and infectious diseases. The Company's diverse pipeline includes its lead program eteplirsen, for Duchenne muscular dystrophy, as well as potential treatments for some of the world's most lethal infectious diseases. Sarepta aims to build a leading, independent biotech company dedicated to translating its RNA-based science into transformational therapeutics for patients who face significant unmet medical needs. For more information, please visit us at www.sarepta.com.
Forward-Looking Statements and Information
This press release contains forward-looking statements. These forward-looking statements generally can be identified by the use of words such as “believes or belief,” “anticipates,” “plans,” “expects,” “will,” “intends,” “potential,” “possible,” “advance” and similar expressions. These forward-looking statements include statements about the development of eteplirsen and its efficacy, potency and utility as a potential treatment for DMD, the potential for the ongoing creation of novel dystrophin and its ability to lead to significant clinical benefit, including as measured by the 6MWT and exploratory measures such as pulmonary function tests, over a longer course of treatment; the potential timing of an NDA submission for eteplirsen in the treatment of DMD; the potential filing and acceptance of an NDA for eteplirsen by the FDA; the timing and submission of additional data, analysis and other information to the FDA necessary for the FDA to make regulatory determinations; the timing and design of and ability to initiate additional studies for eteplirsen; the potential regulatory approval of eteplirsen and the confirmatory trials that may be required in connection with such approval.
Each forward-looking statement contained in this press release is subject to risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statement. Applicable risks and uncertainties include, among others: subsequent clinical trials may fail to demonstrate safety and efficacy of eteplirsen or replicate results; treatment of patients with DMD using eteplirsen over a longer duration may not lead to significant clinical benefit, including as measured by the 6MWT and exploratory measures such as pulmonary function tests; any of Sarepta's drug candidates, including eteplirsen, may fail in development, may not receive required regulatory approvals, or may not become commercially viable during projected time frames or at all due to delays or other reasons; we may not be able to comply with all FDA requests; the FDA may determine that substantial additional data is required for accelerated or other approval of eteplirsen or that our NDA submission for eteplirsen does not qualify for filing, even with additional information; the results of our ongoing and new clinical trials may not be positive; there may be delays in timelines relating to an NDA submission, initiating clinical trials, or making a product commercially available for regulatory or internal reasons; we may not be able to manufacture sufficient supply for clinical trials or commercialization; Agency or court decisions with respect to our patents may negatively impact our business; and those identified under the heading “Risk Factors” in Sarepta’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 filed with the Securities and Exchange Commission (SEC), and Sarepta’s other filings with the SEC.
Any of the foregoing risks could materially and adversely affect Sarepta’s business, results of operations and the trading price of Sarepta’s common stock. We caution investors not to place considerable reliance on the forward-looking statements contained in this press release. Sarepta does not undertake any obligation to publicly update its forward-looking statements based on events or circumstances after the date hereof.
References:
1 ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six minute walk test. Am J Respir Crit Care Med. 2002 Jul 1;166(1): 111 7.
2 Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002 Mar 21;346(12):896 903. Erratum in: N Engl J Med. 2002 Apr 18;346(16):1258.
3 Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double blinded, placebo controlled, multinational study of recombinant human alpha L iduronidase (laronidase). J Pediatr. 2004 May;144(5):581 8.
4 Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM et al.. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006 Aug;8(8):465 73. Erratum in: Genet Med. 2006 Sep;8(9):599.
5 McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL, et al. The 6 minute walk test in Duchenne/Becker muscular dystrophy: longitudinal observations. Muscle Nerve. 2010b Dec;42(6):966 74.
6 Finder J.D., D. Birnkrant, J. Carl, et al. Respiratory care of the patient with Duchenne muscular dystrophy: an official ATS consensus statement. Am J Respir Crit Care Med. 2004; 170: 456–65.
7 Lynn, D. J., R. P. Woda and J. R. Mendell. Respiratory dysfunction in muscular dystrophy and other myopathies. Clin Chest Med. 1994; 15(4): 661-674.
8 Hahn, A., J. R. Bach, A. Delaubier, et al. Clinical implications of maximal respiratory pressure determinations for individuals with Duchenne muscular dystrophy. Arch Phys Med Rehabil
9 McDonald, C. M., R. T. Abresch, G. T. Carter, et al. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995; 74(5 Suppl): S70-92.

Sarepta Media and Investor Contact:
Jim Baker
617.274.4010
jbaker@sarepta.com@mdausa.org

