The Expanding Therapeutic Landscape for Myasthenia Gravis
By MDA Staff | Friday, August 14, 2020
Myasthenia gravis (MG) is an autoimmune disease that leads to muscle weakness. It affects about 14 to 20 out of every 100,000 people. The age of onset varies widely, but typically it is diagnosed in women in their 20s and 30s and men in their 60s or later. In about 10% to 15% of cases, MG begins in childhood.
To learn about the latest MG treatment advances, we talked with Gil I. Wolfe, MD, Irvin and Rosemary Smith professor and chairman of the Department of Neurology at the Jacobs School of Medicine & Biomedical Sciences at the University at Buffalo in New York, who led an extension study on the surgical treatment for MG that was published in The Lancet Neurology in 2019.
What is MG?
It is the most common defect of neuromuscular transmission, or the messaging between nerves and muscles. That disrupted communication leads to muscle weakness, which is why the name means “grave muscular weakness.”
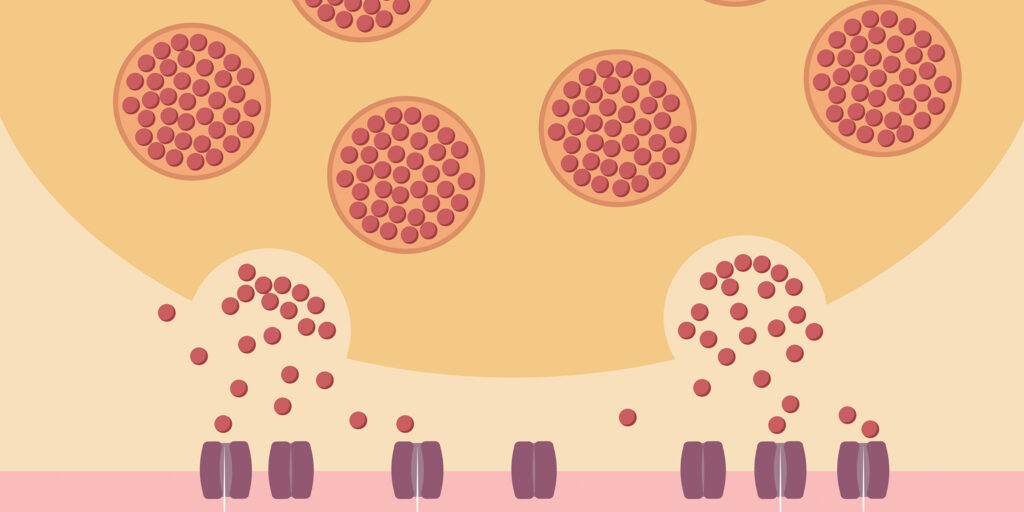
In MG, antibodies (which usually protect us from infections) attack and destroy acetylcholine receptors in the muscles. Nerves release acetylcholine at the neuromuscular junction (where the nerves and muscles intersect). Acetylcholine then binds to receptors on the muscle membrane, much like a key entering a lock. This activates the muscle and allows it to contract. Without enough unlocked receptors, the muscle can’t contract.
The first symptoms of MG typically are eye muscle weakness, double vision, and droopy eyelids. In some patients, weakness around the eyes is the only sign of the disease; we call that ocular MG. The other type is general MG, which also involves muscles in the arms, legs, and chest, including those that control breathing.
What is the surgical treatment for MG?
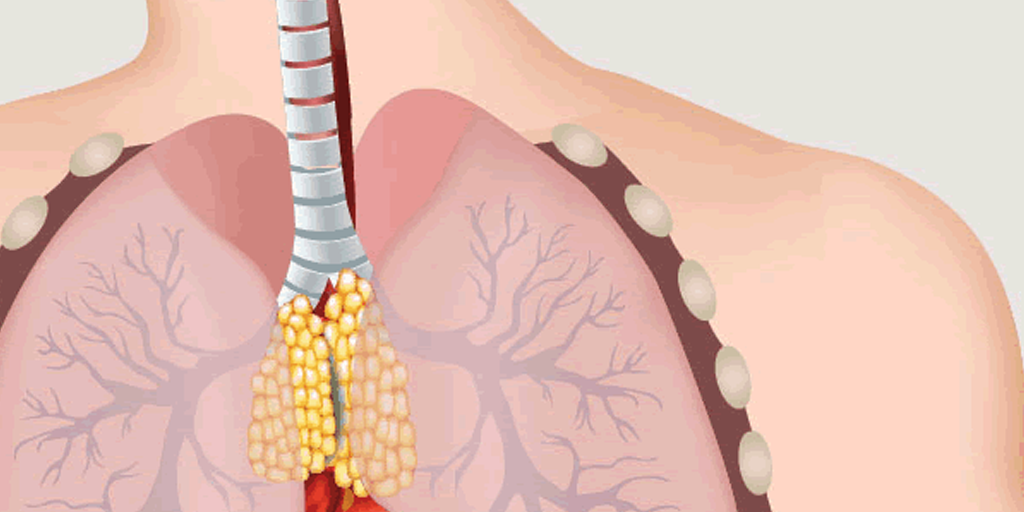
The close association between MG and thymic abnormalities has been known for more than a century. For instance, patients with MG are more likely to get tumors of the thymus. About 80 years ago, observations first appeared in medical literature documenting that removing the thymus gland (a procedure called thymectomy), even if there was no tumor, seemed to benefit patients with MG. But they were small case studies that did not meet current clinical trial standards.
Still, based on those publications, thymectomy became a common treatment for MG. However, doubts remained as to exactly how effective it was in MG patients without thymic tumors.

Gil I. Wolfe, MD
The MGTX thymectomy study, supported by MDA and the National Institutes of Health, was designed to fill that gap in knowledge. The trial compared the effects of thymectomy plus prednisone (a corticosteroid) to prednisone alone. Results showed that removing the thymus in patients with MG improved their disease over a three-year period. They had better muscle control, needed fewer immunosuppressants, and had fewer hospitalizations than those who received prednisone alone.
The MGTX extension study then followed about half the participants for another two years and found the benefits continued.
Most patients with MG won’t reach remission, which is defined as a medication-free year, but we can get patients to minimal manifestation status. This means they can work and manage family obligations and the disease is not notably impairing their function. At the end of the extension study, almost 90% of patients who had the surgery were at minimal manifestation status compared to less than 60% of those who received only medication.
There also appears to be a cost benefit to thymectomy, which is less expensive than emerging therapies or a lifetime of medications. It is invasive, but we now have minimally invasive surgical techniques that are easier on patients.
Will the study change how the disease is treated?
Yes, we are now updating guidelines for the treatment of MG. Also, the American Academy of Neurology released a practice statement on thymectomy, noting that it is safe and effective, and that patients should be aware of the option.
How is MG treated with medication?
We start with an acetylcholinesterase inhibitor like pyridostigmine. These drugs work by keeping more acetylcholine around the neuromuscular junction, thus helping improve nerve/muscle communication and reducing the weakness patients experience. Most people with MG also require immunosuppressants, such as corticosteroids, to suppress the autoimmune response that causes the disease. But these drugs have serious side effects, especially when taken long term, so the goal is to use them sparingly.
We also use IV gamma globulin and plasma exchange when patients have a flareup or if their symptoms don’t improve with other therapies. About 15% of patients don’t respond to conventional treatments.
In 2017, the US Food and Drug Administration (FDA) approved eculizumab (Soliris) for MG, a humanized monoclonal antibody that blocks the complement system, which we know plays a key role in the destruction of the muscle membrane.
What else is under investigation?
The therapeutic investigational landscape in MG is as large and active as it has ever been, and there is a lot of excitement. In addition to anti-B cell therapies (the family of white blood cells that make antibodies) like rituximab, researchers are exploring other monoclonal antibodies, including ravulizumab — which requires less frequent infusions than eculizumab — and zilucoplan, which can be delivered subcutaneously rather than infused. Each of these agents works on the complement system.
Ongoing clinical trials of molecules that block the neonatal fc receptor, which dramatically reduces the amount of Immunoglobulin G antibodies, also show promise.
TAGS: Drug Development, Research, Research Advances, Spotlight
TYPE: Featured Article
Disclaimer: No content on this site should ever be used as a substitute for direct medical advice from your doctor or other qualified clinician.